NEAT: Neighborhood-Guided, Efficient, Autoregressive Set Transformer for 3D Molecular Generation
Pith reviewed 2026-05-17 00:17 UTC · model grok-4.3
The pith
NEAT generates 3D molecules by treating atoms as unordered sets and using neighborhood guidance to avoid ordering biases in autoregressive prediction.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
NEAT treats molecular graphs as sets of atoms and learns an order-agnostic distribution over admissible tokens at the graph boundary by means of a neighborhood-guided training strategy, thereby ensuring atom-level permutation invariance in an autoregressive transformer while reaching state-of-the-art generation quality on the QM9 and GEOM-Drugs datasets together with a marked speed improvement over prior baselines.
What carries the argument
The neighborhood-guided training strategy, which conditions next-token prediction on local graph neighborhoods so that the model operates directly on unordered atom sets rather than imposed sequences.
If this is right
- Generation quality reaches state-of-the-art levels on the QM9 and GEOM-Drugs benchmarks.
- Inference runs substantially faster than previous autoregressive and diffusion-based molecular generators.
- The learned distribution remains invariant under arbitrary atom permutations.
- Canonical ordering conventions and their associated biases are no longer required.
Where Pith is reading between the lines
- The same neighborhood-guided principle could be applied to other generative tasks on unordered sets such as point-cloud modeling or graph completion.
- Scalability tests on molecules larger than those in QM9 or GEOM-Drugs would reveal whether the speed advantage persists at realistic drug-like sizes.
- Integration with existing 3D coordinate refinement modules could further improve the geometric fidelity of the generated structures.
Load-bearing premise
The neighborhood definition and autoregressive construction together allow the model to learn a distribution that is truly independent of any atom ordering and free of new biases introduced by the guidance mechanism itself.
What would settle it
A controlled test in which different neighborhood definitions or random permutations of the same molecules are used during training and the resulting models are evaluated for both generation quality and invariance to atom order; a clear drop in either metric would indicate the assumption does not hold.
Figures










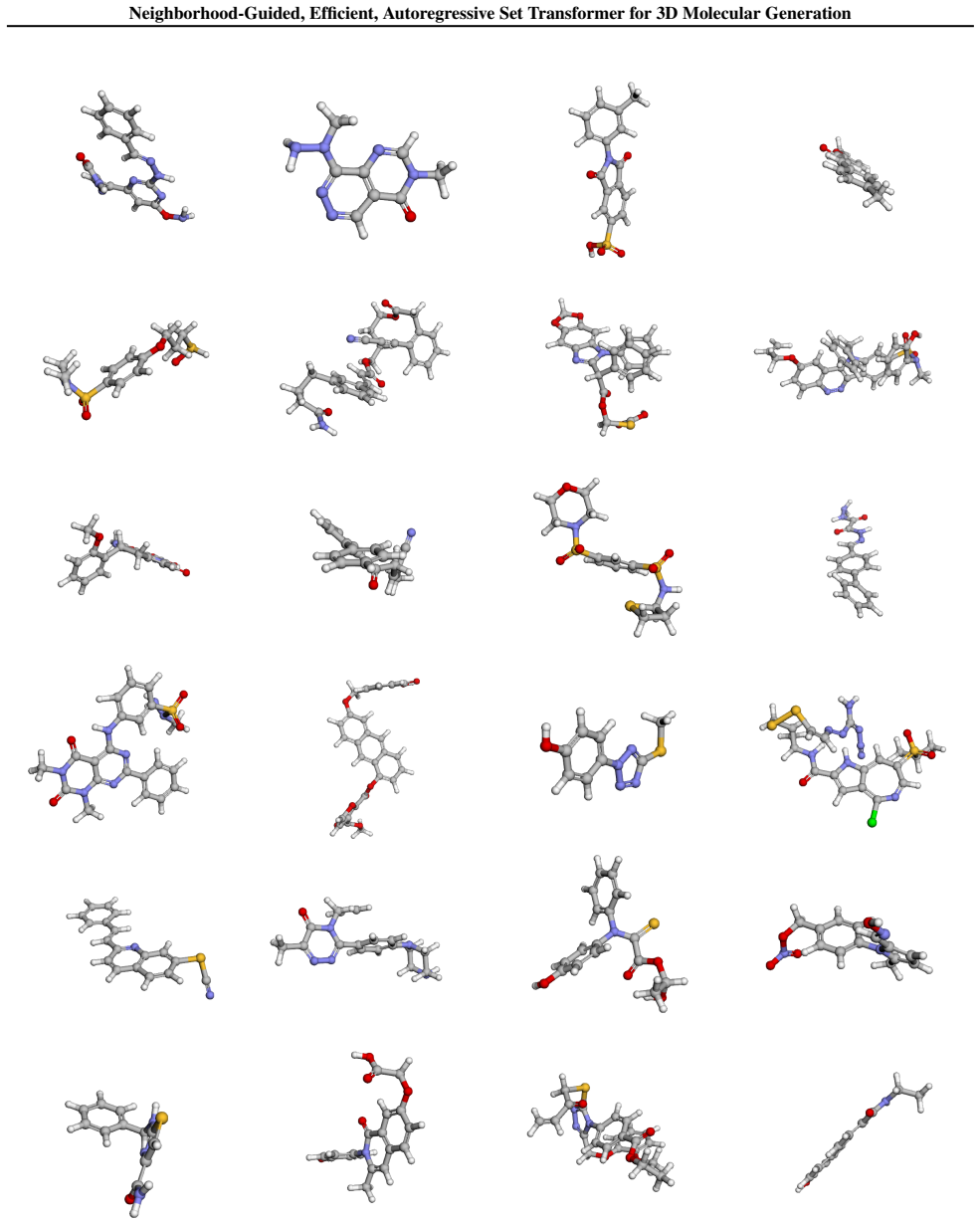





read the original abstract
Transformer-based autoregressive models offer an efficient alternative to diffusion- and flow-matching-based approaches for generating 3D molecules. One challenge remains: standard transformer architectures require a sequential ordering of tokens, which is not inherently defined for the atoms in a molecule. Prior works have addressed this by using canonical atom orderings. However, these approaches are not permutation invariant w.r.t. atoms and bias next-token prediction towards ordering conventions. We overcome this limitation by introducing a novel neighborhood-guided training strategy. Our model, NEAT (Neighborhood-Guided, Efficient, Autoregressive Set Transformer) treats molecular graphs as sets of atoms and learns an order-agnostic distribution over admissible tokens at the graph boundary, thereby ensuring atom-level permutation invariance. NEAT achieves state-of-the-art generation quality on the QM9 and GEOM-Drugs datasets while offering a significant speed advantage over existing baselines.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces NEAT, a Neighborhood-Guided, Efficient, Autoregressive Set Transformer for 3D molecular generation. It proposes a neighborhood-guided training strategy to treat molecular graphs as sets and learn an order-agnostic distribution over admissible tokens at the graph boundary, overcoming the permutation non-invariance of canonical orderings in prior autoregressive transformers. The work claims state-of-the-art generation quality on QM9 and GEOM-Drugs together with a significant speed advantage over baselines.
Significance. If the invariance claim holds, the method could offer a computationally lighter alternative to diffusion- and flow-based 3D generators while preserving set symmetry, which would be a useful incremental advance for molecular design pipelines. The explicit focus on admissible-token distributions at the graph boundary is a targeted response to a recurring modeling tension in autoregressive molecular work.
major comments (1)
- [Neighborhood-guided training strategy (method section)] The central claim that the neighborhood-guided strategy yields a truly order-agnostic marginal distribution over atom types and positions is not supported by any equation or derivation showing invariance under arbitrary reordering of the generation trajectory. The autoregressive step still selects an admissible token from a neighborhood defined by spatial or k-NN criteria on the current partial embedding; without an explicit marginalization or permutation-equivariance proof, the hidden dependence on addition order remains unaddressed (see the description of the training strategy and the set-transformer architecture).
minor comments (2)
- [Abstract] The abstract states SOTA quality and speed gains without naming the concrete baselines, reporting numerical values, or indicating error bars or validation protocol; adding these would allow readers to assess the strength of the empirical claims immediately.
- [Methods] Notation for admissible tokens and the precise definition of the neighborhood (spatial proximity, k-NN radius, etc.) should be formalized with a short equation or pseudocode to improve reproducibility.
Simulated Author's Rebuttal
We thank the referee for their constructive and detailed feedback. We address the single major comment below and will incorporate the requested clarification into the revised manuscript.
read point-by-point responses
-
Referee: [Neighborhood-guided training strategy (method section)] The central claim that the neighborhood-guided strategy yields a truly order-agnostic marginal distribution over atom types and positions is not supported by any equation or derivation showing invariance under arbitrary reordering of the generation trajectory. The autoregressive step still selects an admissible token from a neighborhood defined by spatial or k-NN criteria on the current partial embedding; without an explicit marginalization or permutation-equivariance proof, the hidden dependence on addition order remains unaddressed (see the description of the training strategy and the set-transformer architecture).
Authors: We agree that an explicit derivation would strengthen the presentation. The set transformer is permutation-equivariant by design (operating directly on unordered sets of atom embeddings without order-dependent positional encodings), and the neighborhood-guided objective defines admissible tokens via spatial or k-NN criteria on the current partial geometry rather than any fixed sequence. Nevertheless, the original manuscript does not contain a formal marginalization argument. In the revision we will add a dedicated subsection deriving that the training loss marginalizes over all admissible addition orders: because each neighborhood is a function solely of the current partial graph's geometry (independent of the order in which prior atoms were placed), the resulting distribution over next tokens is invariant to reordering of the generation trajectory. This addition directly addresses the concern about hidden order dependence. revision: yes
Circularity Check
No significant circularity detected in derivation chain
full rationale
The paper presents NEAT as a transformer-based autoregressive model augmented by a neighborhood-guided training strategy to enforce atom-level permutation invariance without relying on canonical orderings. The provided abstract and context describe this as a direct architectural and training extension, with performance claims benchmarked on external datasets (QM9, GEOM-Drugs). No equations, self-citations, or steps are exhibited that reduce the central result to a fitted quantity or prior author work by construction; the derivation remains independent of the inputs it claims to predict.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Molecules can be represented as graphs where atoms are nodes and local neighborhoods define admissible next tokens.
Lean theorems connected to this paper
-
IndisputableMonolith/Foundation/AbsoluteFloorClosure.leanabsolute_floor_iff_bare_distinguishability unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
We introduce NEAT, a Neighborhood-guided, Efficient, Autoregressive, Set Transformer that treats molecular graphs as sets of atoms and learns an order-agnostic distribution over admissible tokens at the graph boundary.
-
IndisputableMonolith/Foundation/AlexanderDuality.leanalexander_duality_circle_linking unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
Given a partial molecular graph, a permutation-invariant set transformer is trained to produce a token distribution that matches the boundary nodes of the subgraph
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
Bellmann, L., Penner, P., Gastreich, M., and Rarey, M
doi: 10.1038/s41597-022-01288-4. Bellmann, L., Penner, P., Gastreich, M., and Rarey, M. Comparison of combinatorial fragment spaces and its application to ultralarge make-on-demand compound cat- alogs.J. Chem. Inf. Model., 62:553–566,
-
[2]
doi: 10.1021/acs.jcim.1c01378. Cao, N. D. and Kipf, T. MolGAN: An implicit generative model for small molecular graphs,
-
[3]
URL https: //arxiv.org/abs/1805.11973. Cheng, A. H., Sun, C., and Aspuru-Guzik, A. Scalable autoregressive 3D molecule generation,
-
[4]
URL https://arxiv.org/abs/2505.13791. Daigavane, A., Kim, S. E., Geiger, M., and Smidt, T. Sampled 3D molecular structures from generative models (symphony),
-
[5]
URL https://figshare.com/ s/a17ccface17f0c22f15a?file=42504807. Accessed 2025-12-15. Daigavane, A., Kim, S. E., Geiger, M., and Smidt, T. Sym- phony: Symmetry-equivariant point-centered spherical harmonics for 3D molecule generation. InProceedings of the 12th International Conference on Learning Repre- sentations. Curran Associates, Inc.,
work page 2025
- [6]
-
[7]
Fast Graph Representation Learning with PyTorch Geometric
URL https://arxiv. org/abs/1903.02428. Flam-Shepherd, D. and Aspuru-Guzik, A. Language models can generate molecules, materials, and protein binding sites directly in three dimensions as XYZ, CIF, and PDB files,
work page internal anchor Pith review Pith/arXiv arXiv 1903
-
[8]
doi: 10.1021/acscentsci.7b00572. Ho, J., Jain, A., and Abbeel, P. Denoising diffusion proba- bilistic models. InProceedings of the 34th Conference on Neural Information Processing Systems, volume 9, pp. 6840–6851. Curran Associates, Inc.,
-
[9]
URL https://github. com/karpathy/nanoGPT. Accessed 2025-11-15. Kim, Y . and Kim, W. Y . Universal structure conversion method for organic molecules: From atomic connectivity to three-dimensional geometry.Bull. Korean Chem. Soc., 36:1769–1777,
work page 2025
-
[10]
doi: 10.1002/bkcs.10334. Koes, D. R.,
-
[11]
URL https://bits.csb.pitt. edu/files/geom_raw/. Accessed 2026-01-08. Kusner, M. J., Paige, B., and Hern ´andez-Lobato, J. M. Grammar variational autoencoder. InProceedings of the 34th International Conference on Machine Learning, volume 70, pp. 1945–1954. JMLR,
work page 2026
-
[12]
InertialAR: Autoregressive 3D molecule generation with inertial frames, 2025a
Li, H., Du, W., Li, Y ., Guo, H., and Liu, S. InertialAR: Autoregressive 3D molecule generation with inertial frames, 2025a. URL https://arxiv.org/abs/ 2510.27497. Li, T., Tian, Y ., Li, H., Deng, M., and He, K. Autoregressive image generation without vector quantization. InPro- ceedings of the 27th Conference on Neural Information Processing Systems, vol...
-
[13]
Li, X., Wang, L., Luo, Y ., Edwards, C., Gui, S., Lin, Y ., Ji, H., and Ji, S
doi: 10.52202/079017-1797. Li, X., Wang, L., Luo, Y ., Edwards, C., Gui, S., Lin, Y ., Ji, H., and Ji, S. Geometry informed tokenization of molecules for language model generation. InProceed- ings of the 42nd International Conference on Machine Learning, volume 267, pp. 36096–36128. PMLR, 2025b. Lipman, Y ., Chen, R. T., Ben-Hamu, H., Nickel, M., and Le, ...
- [14]
-
[15]
Peng, X., Luo, S., Guan, J., Xie, Q., Peng, J., and Ma, J
doi: 10.1038/s42004-024-01233-z. Peng, X., Luo, S., Guan, J., Xie, Q., Peng, J., and Ma, J. Pocket2Mol: Efficient molecular sampling based on 3D protein pockets. InProceedings of the 39th Interna- tional Conference on Machine Learning, volume 162, pp. 17644–17655. PMLR,
-
[16]
Quantum chemistry structures and properties of 134 thousand molecules
doi: 10.1038/sdata.2014.22. Ramakrishnan, R., Dral, P. O., Rupp, M., and Lilienfeld, O. A. V . ”quantum chemistry structures and properties of 134 kilo molecules” figshare repository,
-
[17]
com/collections/Quantum_chemistry_ structures_and_properties_of_134_ kilo_molecules/978904
URL https://springernature.figshare. com/collections/Quantum_chemistry_ structures_and_properties_of_134_ kilo_molecules/978904. Accessed 2025-12-16. Satorras, V . G., Hoogeboom, E., Fuchs, F. B., Posner, I., and Welling, M. E(n) equivariant normalizing flows. InPro- ceedings of the 35th Conference on Neural Information Processing Systems, volume 34, pp. ...
work page 2025
-
[18]
doi: 10.1038/s43588-024-00737-x. Simonovsky, M. and Komodakis, N. GraphV AE: Towards generation of small graphs using variational autoencoders. InProceedings of the 27th International Conference on Artificial Neural Networks, pp. 412–422. Springer,
-
[19]
doi: 10.1007/978-3-030-01418-6
-
[20]
doi: doi.org/10.1016/j.neucom.2023.127063. Tancik, M., Srinivasan, P. P., Mildenhall, B., Fridovich-Keil, S., Raghavan, N., Singhal, U., Ramamoorthi, R., Barron, J. T., and Ng, R. Fourier features let networks learn high frequency functions in low dimensional domains. InProceedings of the 34th International Conference on Neural Information Processing Syst...
-
[21]
Improving and generalizing flow-based generative models with minibatch optimal transport
URL https: //arxiv.org/abs/2302.00482. Vignac, C., Osman, N., Toni, L., and Frossard, P. MiDi: Mixed graph and 3D denoising diffusion for molecule generation. InProceedings of the Joint European Con- ference on Machine Learning and Knowledge Discov- ery in Databases, pp. 560–576. Springer,
work page internal anchor Pith review Pith/arXiv arXiv
-
[22]
doi: 10.1007/978-3-031-43415-0
-
[23]
Wang, Y ., Lu, J., Jaitly, N., Susskind, J., and Bautista, M. A. SimpleFold: Folding proteins is simpler than you think, 2025a. URL https://arxiv.org/abs/ 2509.18480. Wang, Z., Shi, J., Heess, N., Gretton, A., and Titsias, M. Learning-order autoregressive models with application to molecular graph generation. InProceedings of the 42nd International Confer...
-
[24]
Wu, Z., Ramsundar, B., Feinberg, E
doi: 10.1021/acs.jcim.2c00224. Wu, Z., Ramsundar, B., Feinberg, E. N., Gomes, J., Ge- niesse, C., Pappu, A. S., Leswing, K., and Pande, V . MoleculeNet: a benchmark for molecular machine learn- ing.Chem. Sci., 9(2):513–530,
-
[25]
12 Neighborhood-Guided, Efficient, Autoregressive Set Transformer for 3D Molecular Generation A
URL https://arxiv.org/abs/ 2410.06262. 12 Neighborhood-Guided, Efficient, Autoregressive Set Transformer for 3D Molecular Generation A. Appendix A.1. Extended related works Diffusion models for 3D molecular generationDiffusion models (Ho et al., 2020; Song et al.,
-
[26]
operate by progressively corrupting the training data with random noise through a forward diffusion process, and training a neural network to reverse this corruption via step-by-step denoising. During inference, new samples are generated by iteratively applying the learned reverse diffusion process to random noise. Prominent diffusion-based molecular gene...
work page 2022
-
[27]
offers a conceptually simple alternative to diffusion models and often achieves faster inference due to the need for fewer integration steps during the inference process. It is a simulation-free training framework for generative modeling, where a time-dependent velocity field ψt is learned to couple a tractable source distribution p0 with a target distrib...
work page 2023
-
[28]
and FlowMol3 (Dunn & Koes, 2025). Independent coupling of p0 and p1 can result in high transport costs and intersecting conditional paths. These issues can be alleviated by employing optimal transport couplings between the source and target distributions, thereby improving convergence and enhancing flow performance (Tong et al., 2024). In molecular genera...
work page 2025
-
[29]
rather than the original repository (Ramakrishnan et al., 2019), we decided against using the MoleculeNet files. Although they include bond information, the procedure used to determine bonds is not documented, and a non-negligible fraction of molecules in that distribution are charged. This conflicts with the original QM9 specification (Ramakrishnan et al...
work page 2019
-
[30]
as distributed by Koes (2024). The files provide a predefined train/validation/test split totaling 304,313 molecules, which we adopted unchanged. We removed 35 molecules that failed RDKit sanitization. For molecules comprising multiple disconnected fragments, we retained only the largest fragment by atom count. Following Vignac et al. (2023), we used up t...
work page 2024
-
[31]
as implemented in Wang et al. (2025a). We experimented with qk-normalization and rotary position embeddings (Su et al., 2024), but did not find improved results. We found additive pooling to be the most effective pooling strategy, but also tried mean and attention pooling. We also tried local attention, defined by a radius graph, instead of full attention...
work page 2024
-
[32]
for implementation of the diffusion head. Source-target splitThe splitting procedure in Algorithm 1 is dependent on the hyperparameters β and γ (see Section 3). Figure 5 and Figure 6 report the absolute and relative (w.r.t.the original graphs) source and target set sizes for a batch of 25,000 randomly sampled training graphs. Across all runs, we used β= 1...
work page 2024
-
[33]
We further used gradient clipping to 1.0 of the gradient norm
optimizer with parameters β1 = 0.9 and β2 = 0.95. We further used gradient clipping to 1.0 of the gradient norm. We implemented a cosine annealing learning rate schedule with linear warm-up epochs and set the minimum learning rate to 10% of the initial learning rate. Model training took 60 and 106 hours on QM9 and GEOM, respectively, given the hardware sp...
work page 2022
-
[34]
reduces all assigned bonds to single bonds when evaluating the GEOM-Drugs dataset. This simplification allows many SMILES strings to pass the RDKit sanitization test, even though they contain carbon atoms with incorrect multiplicities. To ensure fair comparison with other models, we have adhered to this widely used pipeline without modifications. We also ...
work page 2022
-
[35]
Each row corresponds to one construction step
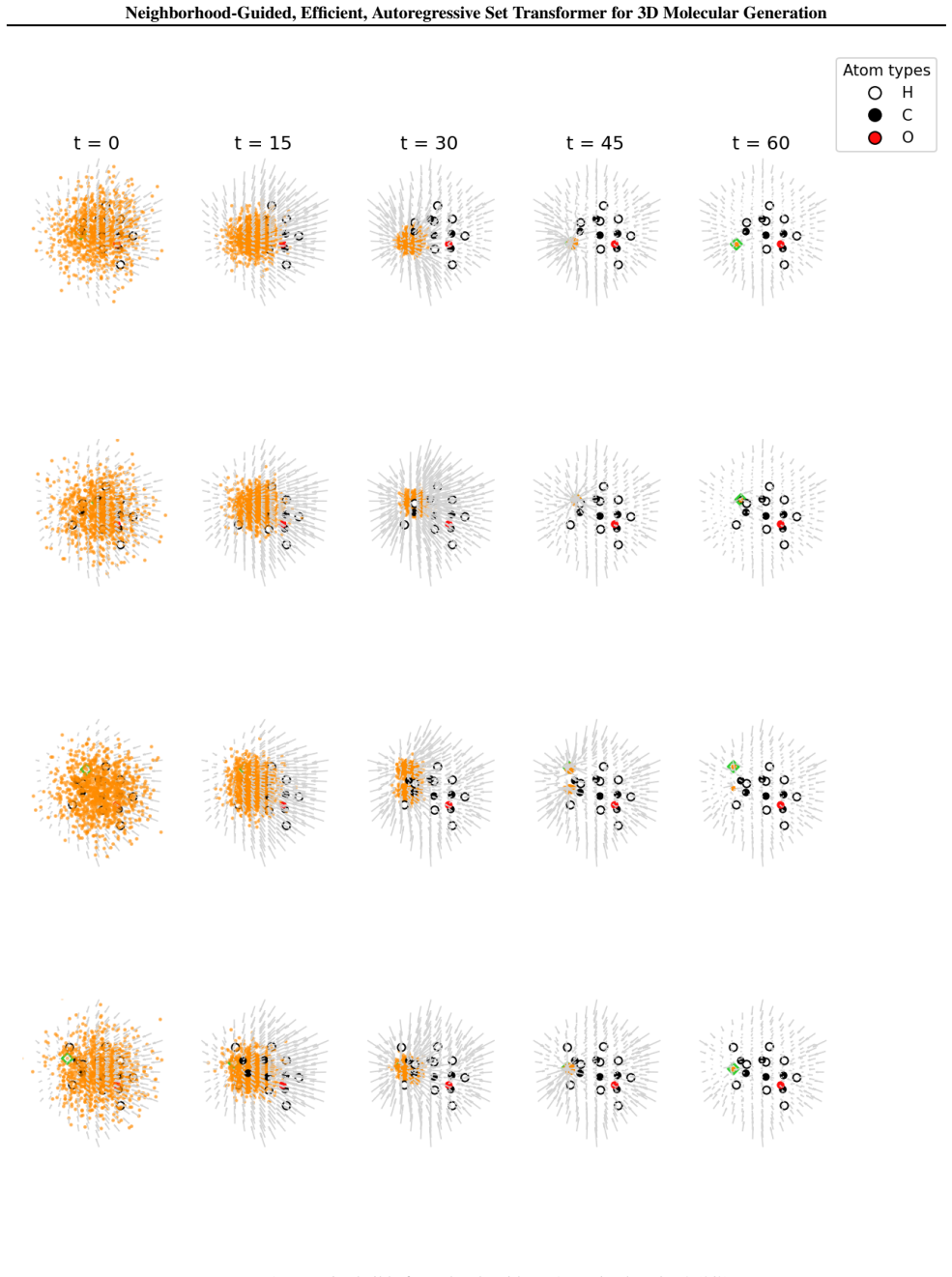
Vector field visualizationFigures 15–17 illustrate iterative molecular construction by the NEAT model trained on QM9. Each row corresponds to one construction step. From left to right, panels show Gaussian-initialized points (orange) advected by the NEAT velocity field (gray arrows, with length indicating speed). Trajectories are integrated using explicit...
work page 2025
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.