SAVE: A Generalizable Framework for Multi-Condition Single-Cell Generation with Gene Block Attention
Pith reviewed 2026-05-10 07:42 UTC · model grok-4.3
The pith
SAVE demonstrates that grouping genes into semantic blocks enables more accurate generation of single-cell profiles for previously unseen combinations of conditions.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
SAVE is a unified generative framework based on conditional Transformers for multi-condition single-cell modeling. It introduces a coarse-grained representation by grouping semantically related genes into blocks, thereby capturing higher-order dependencies among gene modules rather than treating genes as independent tokens. A Flow Matching mechanism combined with a condition-masking strategy supports flexible simulation and generalization to unseen condition combinations. When tested on benchmarks for conditional generation, batch effect correction, and perturbation prediction, SAVE achieves higher generation fidelity and better extrapolative performance than state-of-the-art alternatives,特に
What carries the argument
Gene block attention, which partitions genes into blocks of semantically related genes to model collective patterns and higher-order dependencies instead of independent gene tokens.
If this is right
- More reliable simulation of cellular responses to novel mixes of perturbations or environments not present in training data.
- Improved correction of technical batch effects when integrating single-cell datasets collected under different conditions.
- Stronger performance on perturbation prediction tasks when only partial condition data is available.
- Lower data requirements for exploring large spaces of possible condition combinations in virtual cell modeling.
Where Pith is reading between the lines
- The block-grouping idea may transfer to other high-dimensional biological datasets that exhibit modular structure, such as spatial or multi-omics profiles.
- Condition masking could be adapted to generative tasks in other domains where models must handle partially observed input factors.
- Examining which gene blocks respond most strongly to particular conditions might yield testable biological hypotheses about regulatory modules.
Load-bearing premise
That grouping genes into semantic blocks captures the higher-order biological dependencies sufficiently well to support accurate generation and true extrapolation to new condition combinations.
What would settle it
Hold out all data from one specific combination of conditions during training, generate synthetic single-cell profiles for that exact combination, and check whether key statistics such as gene correlations and expression distributions match real measured cells from the same combination better than baseline models.
Figures











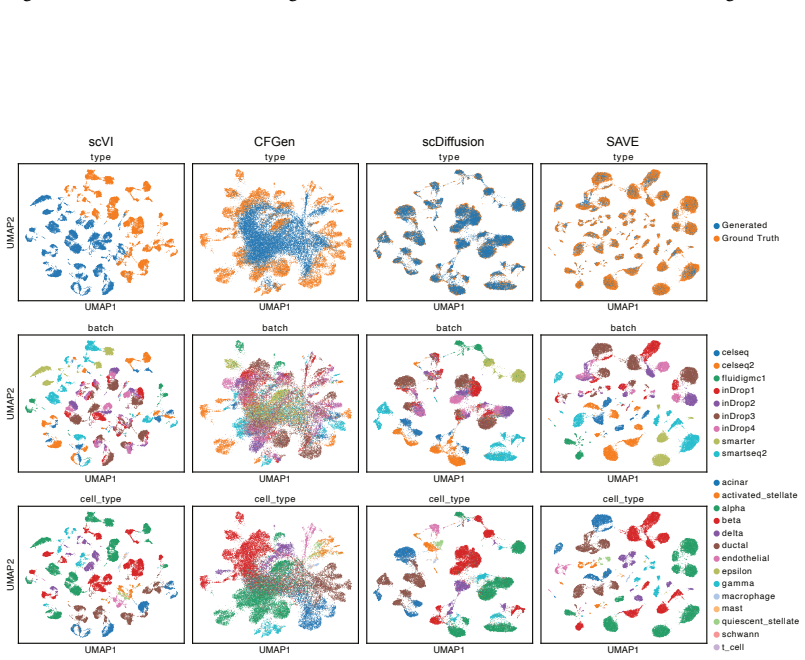


read the original abstract
Modeling single-cell gene expression across diverse biological and technical conditions is crucial for characterizing cellular states and simulating unseen scenarios. Existing methods often treat genes as independent tokens, overlooking their high-level biological relationships and leading to poor performance. We introduce SAVE, a unified generative framework based on conditional Transformers for multi-condition single-cell modeling. SAVE leverages a coarse-grained representation by grouping semantically related genes into blocks, capturing higher-order dependencies among gene modules. A Flow Matching mechanism and condition-masking strategy further enhance flexible simulation and enable generalization to unseen condition combinations. We evaluate SAVE on a range of benchmarks, including conditional generation, batch effect correction, and perturbation prediction. SAVE consistently outperforms state-of-the-art methods in generation fidelity and extrapolative generalization, especially in low-resource or combinatorially held-out settings. Overall, SAVE offers a scalable and generalizable solution for modeling complex single-cell data, with broad utility in virtual cell synthesis and biological interpretation. Our code is publicly available at https://github.com/fdu-wangfeilab/sc-save
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces SAVE, a conditional Transformer framework for multi-condition single-cell gene expression modeling. It groups semantically related genes into blocks to capture higher-order dependencies, employs Flow Matching for flexible generation, and uses a condition-masking strategy to support simulation of unseen condition combinations. The paper evaluates the model on conditional generation, batch effect correction, and perturbation prediction benchmarks, claiming consistent outperformance over state-of-the-art methods in fidelity and extrapolative generalization, especially under low-resource or combinatorially held-out conditions. Code is released publicly.
Significance. If the empirical claims hold after addressing the extrapolation verification gap, SAVE would represent a meaningful advance in single-cell generative modeling by incorporating biological module structure and enabling more reliable simulation of novel condition sets. This could support virtual cell applications and perturbation studies. Public code availability is a clear strength for reproducibility.
major comments (2)
- [Results (held-out and perturbation benchmarks)] Results section on combinatorially held-out settings and perturbation prediction: the central claim of extrapolative generalization via condition-masking lacks any analysis showing that held-out condition vectors lie outside the span of training correlations (e.g., no linear reconstruction test or shared-factor ablation). Without this, reported gains may reflect interpolation on co-occurrence patterns rather than true extrapolation, directly undermining the generalizability assertions.
- [Method (Gene Block Attention)] Method section describing gene-block construction and attention: no ablation isolates the contribution of prior-knowledge semantic blocks versus learned groupings or standard per-gene tokenization. This component is load-bearing for the claim that block attention captures higher-order dependencies missed by existing methods.
minor comments (2)
- [Abstract] Abstract: quantitative metrics, error bars, and exact baseline comparisons are asserted but not reported; the full results tables should be referenced here for immediate clarity.
- [Method] Notation for Flow Matching and masking probabilities could be unified with a single equation reference to avoid ambiguity across sections.
Simulated Author's Rebuttal
We thank the referee for their detailed and constructive review of our manuscript. We address each of the major comments below and outline the revisions we plan to make to strengthen the paper.
read point-by-point responses
-
Referee: Results section on combinatorially held-out settings and perturbation prediction: the central claim of extrapolative generalization via condition-masking lacks any analysis showing that held-out condition vectors lie outside the span of training correlations (e.g., no linear reconstruction test or shared-factor ablation). Without this, reported gains may reflect interpolation on co-occurrence patterns rather than true extrapolation, directly undermining the generalizability assertions.
Authors: We agree that additional verification is needed to confirm that the held-out condition vectors represent true extrapolation. In the revised manuscript, we will incorporate a linear reconstruction test and a shared-factor ablation analysis. These will demonstrate that the held-out conditions lie outside the span of training correlations, thereby supporting that the observed gains stem from the condition-masking strategy's ability to generalize to novel combinations rather than mere interpolation. revision: yes
-
Referee: Method section describing gene-block construction and attention: no ablation isolates the contribution of prior-knowledge semantic blocks versus learned groupings or standard per-gene tokenization. This component is load-bearing for the claim that block attention captures higher-order dependencies missed by existing methods.
Authors: We acknowledge that an ablation study is necessary to isolate the impact of using prior-knowledge semantic blocks. We will add such ablations in the revision, comparing the performance of our gene block attention against versions using learned groupings and standard per-gene tokenization. This will provide evidence that the semantic blocks contribute to capturing higher-order dependencies that are not addressed by alternative approaches. revision: yes
Circularity Check
No significant circularity detected
full rationale
The SAVE framework introduces a conditional Transformer architecture with gene-block grouping, Flow Matching, and condition-masking for single-cell generation. All performance claims rest on external benchmark evaluations (conditional generation, batch correction, perturbation prediction) against held-out data and prior SOTA methods, with no equations or results shown to reduce by construction to quantities fitted on the evaluation sets themselves. No load-bearing self-citations, uniqueness theorems, or ansatzes imported from prior author work are invoked to justify the central generalization results. The derivation chain is self-contained against independent benchmarks.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Genes can be grouped into semantically related blocks that capture higher-order dependencies.
invented entities (1)
-
Gene blocks
no independent evidence
Reference graph
Works this paper leans on
-
[1]
" write newline "" before.all 'output.state := FUNCTION n.dashify 't := "" t empty not t #1 #1 substring "-" = t #1 #2 substring "--" = not "--" * t #2 global.max substring 't := t #1 #1 substring "-" = "-" * t #2 global.max substring 't := while if t #1 #1 substring * t #2 global.max substring 't := if while FUNCTION format.date year duplicate empty "emp...
-
[2]
\@ifxundefined[1] #1\@undefined \@firstoftwo \@secondoftwo \@ifnum[1] #1 \@firstoftwo \@secondoftwo \@ifx[1] #1 \@firstoftwo \@secondoftwo [2] @ #1 \@temptokena #2 #1 @ \@temptokena \@ifclassloaded agu2001 natbib The agu2001 class already includes natbib coding, so you should not add it explicitly Type <Return> for now, but then later remove the command n...
-
[3]
\@lbibitem[] @bibitem@first@sw\@secondoftwo \@lbibitem[#1]#2 \@extra@b@citeb \@ifundefined br@#2\@extra@b@citeb \@namedef br@#2 \@nameuse br@#2\@extra@b@citeb \@ifundefined b@#2\@extra@b@citeb @num @parse #2 @tmp #1 NAT@b@open@#2 NAT@b@shut@#2 \@ifnum @merge>\@ne @bibitem@first@sw \@firstoftwo \@ifundefined NAT@b*@#2 \@firstoftwo @num @NAT@ctr \@secondoft...
-
[4]
N 3" F/JPrb[䥟 Qd [Sl1x #bG 3I [ql2 8x t rp/8 p Cfq .Knjm͠ r28 ?.)ɩL^6g, qm
@open @close @open @close and [1] URL: #1 \@ifundefined chapter * \@mkboth \@ifxundefined @sectionbib * \@mkboth * \@mkboth\@gobbletwo \@ifclassloaded amsart * \@ifclassloaded amsbook * \@ifxundefined @heading @heading NAT@ctr thebibliography [1] @ \@biblabel @NAT@ctr \@bibsetup #1 @NAT@ctr @ @openbib .11em \@plus.33em \@minus.07em 4000 4000 `\.\@m @bibit...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.