An Oracle-Free Quantum Algorithm for Nonadiabatic Quantum Molecular Dynamics
Pith reviewed 2026-05-10 02:39 UTC · model grok-4.3
The pith
An oracle-free quantum algorithm simulates nonadiabatic molecular dynamics by applying diabatic Hamiltonians directly via first-quantized split-operator propagators.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Nonadiabatic quantum molecular dynamics can be simulated on quantum hardware by applying diabatic Hamiltonian operators directly to the computational basis as first-quantized split-operator propagators, validated through dynamic observables including absorption and recurrence spectra, scattering cross-sections, population dynamics, and quantum scars, with circuit optimizations for multi-mode extensions and demonstrated advantages in depth and T-count.
What carries the argument
First-quantized split-operator propagator for diabatic Hamiltonians, which decomposes time evolution into kinetic and potential operators implementable directly on the qubit basis without oracles.
If this is right
- Enables circuit optimization for multi-mode and multi-channel cases using multivariate potentials and graph-theoretic reductions from molecular symmetry.
- Yields circuit depth advantage over QROM-loading architectures on fault-tolerant scales.
- Retains scalable T-gate advantage for the Trotter architecture relative to quantum signal processing variants.
- Extends via finite-basis and discrete-variable duality to congruent circuit decompositions for dynamics beyond electronic states.
Where Pith is reading between the lines
- The direct diabatic implementation may generalize to other parameterized Hamiltonians in physics that resist standard oracle or diagonalization techniques.
- Symmetry-based graph optimizations could reduce resources for simulating symmetric quantum systems outside chemistry.
- Early verification on small molecular test cases would confirm whether the projected T-gate savings appear before large-scale hardware is available.
- The method opens a route to modeling photochemical processes involving multiple coupled electronic surfaces on quantum processors.
Load-bearing premise
Diabatic Hamiltonian operators can be implemented directly on the computational basis with efficient first-quantized split-operator propagators without hidden oracle costs or significant approximation errors in multi-mode and multi-channel extensions.
What would settle it
Implement the circuit for a small two-mode nonadiabatic system such as a diatomic molecule, compute population dynamics or absorption spectrum, and compare resource counts and accuracy against both exact classical results and a QROM-based reference; mismatch in T-gate scaling or excess Trotter error would falsify the advantage claim.
Figures






























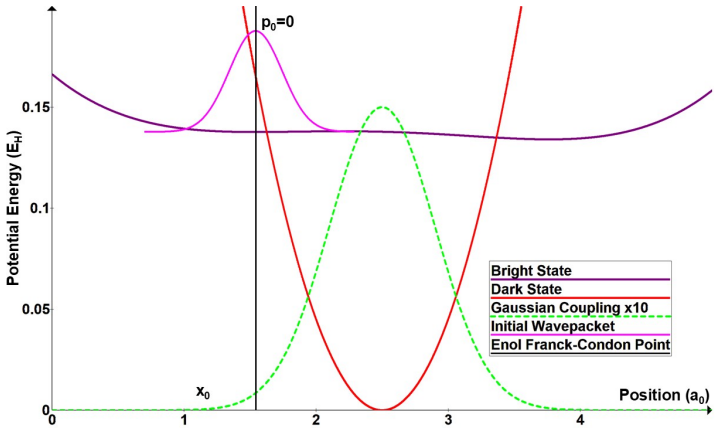







read the original abstract
Quantum computation is an attractive front for many problems that are intractable for computers today. One such problem is nonadiabatic quantum molecular dynamics, where quantized internal states coupling to parameterized modes result in a Hamiltonian resistant to oracle-based models and spectral decomposition. This dissertation applies diabatic Hamiltonian operators directly to the computational basis as first-quantized split-operator propagators, validated with dynamic observables including absorption and recurrence spectra, scattering cross-sections, population dynamics, and quantum scars. Circuits are derived and specified, with focused circuit optimization in multi-mode and multi-channel extensions, including multivariate potential energy terms and graph theoretic optimization from molecular symmetry. Resource estimation shows circuit depth advantage against QROM-loading architectures on a fault-tolerant scale, and a quantitative comparison against quantum signal processing variants confirms that a Trotter-based architecture retains a scalable T-gate advantage. Expanding beyond electronic states demonstrates that duality between finite basis and discrete variable representations permits congruent structural decompositions into quantum circuits, expanding the use of multi-channel dynamics far beyond chemistry.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes an oracle-free quantum algorithm for nonadiabatic quantum molecular dynamics that applies diabatic Hamiltonian operators directly via first-quantized split-operator propagators on the computational basis. It validates the approach through dynamic observables (absorption/recurrence spectra, scattering cross-sections, population dynamics, quantum scars), derives and optimizes circuits for multi-mode/multi-channel extensions including multivariate potentials and symmetry reductions, and reports resource estimates claiming circuit-depth and T-gate advantages over QROM-loading architectures and quantum signal processing variants. The work also extends the framework via finite-basis/discrete-variable duality to broader multi-channel dynamics.
Significance. If the claimed resource advantages are substantiated by explicit gate counts and error bounds, the approach could enable scalable fault-tolerant simulation of nonadiabatic dynamics without oracle assumptions, offering a concrete alternative to spectral or QSP methods for systems with coupled modes and channels.
major comments (2)
- [Resource estimation and circuit derivation sections] The central resource-estimation claim (circuit depth and scalable T-gate advantage versus QROM and QSP) rests on the implementation cost of the multivariate potential-energy terms within the multi-mode, multi-channel split-operator propagator. No explicit gate-count breakdown, circuit diagram, or scaling analysis for these operators is provided that would confirm only polynomial overhead without implicit QROM-style loading or channel-dependent Trotter-error growth; this directly affects whether the reported advantage holds.
- [Validation and observables section] Validation is stated via observables, yet no quantitative error analysis, Trotter-error bounds, or convergence data with respect to time-step, basis size, or number of channels is supplied. This leaves the accuracy of the first-quantized diabatic propagator unquantified for the multi-channel extensions that underpin the scalability claims.
minor comments (1)
- Notation for the diabatic Hamiltonian and split-operator decomposition should be introduced with explicit operator definitions before the circuit constructions to improve readability.
Simulated Author's Rebuttal
We thank the referee for their thorough review and valuable feedback on our manuscript. We address each of the major comments below and will make the necessary revisions to strengthen the presentation of our results.
read point-by-point responses
-
Referee: [Resource estimation and circuit derivation sections] The central resource-estimation claim (circuit depth and scalable T-gate advantage versus QROM and QSP) rests on the implementation cost of the multivariate potential-energy terms within the multi-mode, multi-channel split-operator propagator. No explicit gate-count breakdown, circuit diagram, or scaling analysis for these operators is provided that would confirm only polynomial overhead without implicit QROM-style loading or channel-dependent Trotter-error growth; this directly affects whether the reported advantage holds.
Authors: We agree that the resource estimation would be strengthened by more explicit gate-count breakdowns and circuit diagrams for the multivariate potential-energy terms. In the revised manuscript, we will add a detailed breakdown of the gate counts for the first-quantized split-operator implementation of these terms, along with a scaling analysis that confirms polynomial overhead in the multi-mode case. This analysis will explicitly show direct application in the computational basis without any QROM-style loading. We will also provide circuit diagrams for the key operator implementations and extend the T-gate comparison to QSP variants with the updated counts to substantiate the claimed advantage. revision: yes
-
Referee: [Validation and observables section] Validation is stated via observables, yet no quantitative error analysis, Trotter-error bounds, or convergence data with respect to time-step, basis size, or number of channels is supplied. This leaves the accuracy of the first-quantized diabatic propagator unquantified for the multi-channel extensions that underpin the scalability claims.
Authors: The current validation demonstrates agreement with established observables for benchmark systems. To quantify the accuracy more rigorously, the revised manuscript will include explicit Trotter-error bounds for the diabatic split-operator propagator, derived from the standard error analysis for such methods. We will also add convergence data showing dependence on time-step and basis size, and extend this to demonstrate controlled error growth with the number of channels, confirming the suitability for the multi-channel scalability claims. revision: yes
Circularity Check
No circularity: derivation relies on explicit circuit constructions and external comparisons
full rationale
The paper derives first-quantized split-operator circuits for diabatic Hamiltonians applied directly to the computational basis, with optimizations for multi-mode/multi-channel cases including symmetry reductions. Resource estimates and T-gate comparisons to QROM and QSP architectures are presented as quantitative outputs of those constructions. No quoted step equates a claimed prediction or uniqueness result to a fitted input, self-citation, or ansatz by construction; the central claims remain independent of the paper's own fitted values or prior self-references. The derivation chain is therefore self-contained against the stated benchmarks.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption Fault-tolerant quantum computing with sufficient qubits and gates is available for resource estimation
- domain assumption Diabatic Hamiltonian operators can be applied directly without additional oracles or loading costs
Reference graph
Works this paper leans on
-
[1]
The complexity of enumeration and reliability problems.siam Journal on Computing, 8(3):410–421, 1979
Leslie G Valiant. The complexity of enumeration and reliability problems.siam Journal on Computing, 8(3):410–421, 1979
work page 1979
-
[2]
Ivan Kassal, James D Whitfield, Alejandro Perdomo-Ortiz, Man-Hong Yung, and Al´ an Aspuru-Guzik. Simulating chemistry using quantum computers.Annual review of physical chemistry, 62(1):185–207, 2011
work page 2011
-
[3]
Julia Westermayr, Michael Gastegger, Maximilian FSJ Menger, Sebastian Mai, Leticia Gonz´ alez, and Philipp Marquetand. Machine learning enables long time scale molecular photodynamics simulations.Chemical science, 10(35):8100–8107, 2019
work page 2019
-
[4]
Pavlo O Dral, Mario Barbatti, and Walter Thiel. Nonadiabatic excited-state dynamics with machine learning.The journal of physical chemistry letters, 9(19):5660–5663, 2018
work page 2018
-
[5]
Alexey V Akimov. Extending the time scales of nonadiabatic molecular dynamics via machine learning in the time domain.The Journal of Physical Chemistry Letters, 12(50):12119–12128, 2021
work page 2021
-
[6]
Changwei Zhang, Yang Zhong, Zhi-Guo Tao, Xinming Qin, Honghui Shang, Zhenggang Lan, Oleg V Prezhdo, Xin-Gao Gong, Weibin Chu, and Hongjun Xiang. Advancing nonadiabatic molecular dynamics simulations in solids with e (3) equivariant deep neural hamiltonians.Nature Communications, 16(1):2033, 2025. 351
work page 2033
-
[7]
Carolin M¨ uller,ˇStˇ ep´ an Srˇ seˇ n, Brigitta Bachmair, Rachel Crespo-Otero, Jingbai Li, Sascha Mausenberger, Max Pinheiro, Graham Worth, Steven A Lopez, and Julia West- ermayr. Machine learning for nonadiabatic molecular dynamics: best practices and recent progress.Chemical Science, 16(38):17542–17567, 2025
work page 2025
-
[8]
Simon Axelrod, Eugene Shakhnovich, and Rafael G´ omez-Bombarelli. Excited state non-adiabatic dynamics of large photoswitchable molecules using a chemically trans- ferable machine learning potential.Nature communications, 13(1):3440, 2022
work page 2022
-
[9]
Nikolay Kondratyuk, Vsevolod Nikolskiy, Daniil Pavlov, and Vladimir Stegailov. Gpu- accelerated molecular dynamics: State-of-art software performance and porting from nvidia cuda to amd hip.The International Journal of High Performance Computing Applications, 35(4):312–324, 2021
work page 2021
-
[10]
Stefan Seritan, Christoph Bannwarth, Bryan S Fales, Edward G Hohenstein, Chris- tine M Isborn, Sara IL Kokkila-Schumacher, Xin Li, Fang Liu, Nathan Luehr, James W Snyder Jr, et al. Terachem: A graphical processing unit-accelerated electronic structure package for large-scale ab initio molecular dynamics.Wiley Interdisciplinary Reviews: Computational Molec...
work page 2021
-
[11]
Laurens DM Peters, Jorg Kussmann, and Christian Ochsenfeld. Nonadiabatic molec- ular dynamics on graphics processing units: Performance and application to rotary molecular motors.Journal of Chemical Theory and Computation, 15(12):6647–6659, 2019
work page 2019
-
[12]
The deuterium chemistry of the early universe
PC Stancil, S Lepp, and A Dalgarno. The deuterium chemistry of the early universe. The Astrophysical Journal, 509(1):1, 1998
work page 1998
-
[13]
Ryan C Fortenberry. Quantum chemistry and astrochemistry: a match made in the heavens.The Journal of Physical Chemistry A, 128(9):1555–1565, 2024. 352
work page 2024
-
[14]
Rovibrationally resolved photodissociation of heh+.The Astrophysical Journal, 735(1):21, 2011
S Miyake, CD Gay, and PC Stancil. Rovibrationally resolved photodissociation of heh+.The Astrophysical Journal, 735(1):21, 2011
work page 2011
-
[15]
Meilani Wibowo, Maurizio Persico, and Giovanni Granucci. Nonadiabatic dynamics simulations of singlet fission in 2, 5-bis (fluorene-9-ylidene)-2, 5-dihydrothiophene crys- tals.Physical Chemistry Chemical Physics, 21(2):692–701, 2019
work page 2019
-
[16]
Piermichele Kobauri, Frank J Dekker, Wiktor Szymanski, and Ben L Feringa. Ratio- nal design in photopharmacology with molecular photoswitches.Angewandte Chemie International Edition, 62(30):e202300681, 2023
work page 2023
-
[17]
Jimmy K Yu, Christoph Bannwarth, Ruibin Liang, Edward G Hohenstein, and Todd J Mart´ ınez. Nonadiabatic dynamics simulation of the wavelength-dependent photochem- istry of azobenzene excited to the nπ* andππ* excited states.Journal of the American Chemical Society, 142(49):20680–20690, 2020
work page 2020
-
[18]
Ruibin Liang and Amirhossein Bakhtiiari. Effects of enzyme–ligand interactions on the photoisomerization of a light-regulated chemotherapeutic drug.The Journal of Physical Chemistry B, 126(12):2382–2393, 2022
work page 2022
-
[19]
Tammie R Nelson, Alexander J White, Josiah A Bjorgaard, Andrew E Sifain, Yu Zhang, Benjamin Nebgen, Sebastian Fernandez-Alberti, Dmitry Mozyrsky, Adrian E Roitberg, and Sergei Tretiak. Non-adiabatic excited-state molecular dy- namics: Theory and applications for modeling photophysics in extended molecular materials.Chemical reviews, 120(4):2215–2287, 2020
work page 2020
-
[20]
Linjun Wang, Alexey Akimov, and Oleg V Prezhdo. Recent progress in surface hopping: 2011–2015.The journal of physical chemistry letters, 7(11):2100–2112, 2016. 353
work page 2011
-
[21]
Dongyu Liu, Bipeng Wang, Yifan Wu, Andrey S Vasenko, and Oleg V Prezhdo. Break- ing the size limitation of nonadiabatic molecular dynamics in condensed matter sys- tems with local descriptor machine learning.Proceedings of the National Academy of Sciences, 121(36):e2403497121, 2024
work page 2024
-
[22]
Qijing Zheng, Weibin Chu, Chuanyu Zhao, Lili Zhang, Hongli Guo, Yanan Wang, Xiang Jiang, and Jin Zhao. Ab initio nonadiabatic molecular dynamics investigations on the excited carriers in condensed matter systems.Wiley Interdisciplinary Reviews: Computational Molecular Science, 9(6):e1411, 2019
work page 2019
-
[23]
Singlet fission.Chemical reviews, 110(11):6891– 6936, 2010
Millicent B Smith and Josef Michl. Singlet fission.Chemical reviews, 110(11):6891– 6936, 2010
work page 2010
-
[24]
Alexey V Akimov and Oleg V Prezhdo. Nonadiabatic dynamics of charge transfer and singlet fission at the pentacene/c60 interface.Journal of the American Chemical Society, 136(4):1599–1608, 2014
work page 2014
-
[25]
Guohua Tao. Electronically nonadiabatic dynamics in singlet fission: A quasi-classical trajectory simulation.The Journal of Physical Chemistry C, 118(31):17299–17305, 2014
work page 2014
-
[26]
Leon Freitag and Markus Reiher. The density matrix renormalization group for strong correlation in ground and excited states.Quantum chemistry and dynamics of excited states: Methods and applications, pages 205–245, 2020
work page 2020
-
[27]
John C Tully. Molecular dynamics with electronic transitions.The Journal of Chemical Physics, 93(2):1061–1071, 1990
work page 1990
-
[28]
Michael F Herman and Michael P Moody. Numerical study of the accuracy and effi- ciency of various approaches for monte carlo surface hopping calculations.The Journal of chemical physics, 122(9), 2005. 354
work page 2005
-
[29]
Vyacheslav N Gorshkov, Sergei Tretiak, and Dmitry Mozyrsky. Semiclassical monte- carlo approach for modelling non-adiabatic dynamics in extended molecules.Nature communications, 4(1):2144, 2013
work page 2013
-
[30]
Dynamic programming.science, 153(3731):34–37, 1966
Richard Bellman. Dynamic programming.science, 153(3731):34–37, 1966
work page 1966
-
[31]
Eduarda Sangiogo-Gil and Leticia Gonz´ alez. Nonadiabatic molecular dynamics on quantum computers: challenges and opportunities.Pure and Applied Chemistry, 97(11):1647–1665, 2025
work page 2025
-
[32]
Universal quantum simulators.Science, 273(5278):1073–1078, 1996
Seth Lloyd. Universal quantum simulators.Science, 273(5278):1073–1078, 1996
work page 1996
-
[33]
Hamiltonian simulation by qubitization.Quan- tum, 3:163, 2019
Guang Hao Low and Isaac L Chuang. Hamiltonian simulation by qubitization.Quan- tum, 3:163, 2019
work page 2019
-
[34]
Philipp Schleich, Lasse Bjørn Kristensen, Jorge A Campos-Gonzalez-Angulo, Abdul- rahman Aldossary, Davide Avagliano, Mohsen Bagherimehrab, Christoph Gorgulla, Joe Fitzsimons, and Al´ an Aspuru-Guzik. Chemically motivated simulation problems are efficiently solvable on a quantum computer.Digital Discovery, 5(1):64–87, 2026
work page 2026
-
[35]
Guang Hao Low and Isaac L Chuang. Optimal hamiltonian simulation by quantum signal processing.Physical review letters, 118(1):010501, 2017
work page 2017
-
[36]
Andr´ as Gily´ en, Yuan Su, Guang Hao Low, and Nathan Wiebe. Quantum singular value transformation and beyond: exponential improvements for quantum matrix arith- metics. InProceedings of the 51st annual ACM SIGACT symposium on theory of computing, pages 193–204, 2019
work page 2019
-
[37]
Generalized quantum signal processing.PRX Quantum, 5(2):020368, 2024
Danial Motlagh and Nathan Wiebe. Generalized quantum signal processing.PRX Quantum, 5(2):020368, 2024
work page 2024
-
[38]
Experimental quantum simulation of chemical dynamics
Tomas Navickas, Ryan J MacDonell, Christophe H Valahu, Vanessa C Olaya-Agudelo, Frank Scuccimarra, Maverick J Millican, Vassili G Matsos, Henry L Nourse, Arjun D 355 Rao, Michael J Biercuk, et al. Experimental quantum simulation of chemical dynamics. Journal of the American Chemical Society, 2025
work page 2025
-
[39]
Jacob Whitlow, Zhubing Jia, Ye Wang, Chao Fang, Jungsang Kim, and Kenneth R Brown. Quantum simulation of conical intersections using trapped ions.Nature Chem- istry, 15(11):1509–1514, 2023
work page 2023
-
[40]
Pauline Jeanne Ollitrault.Solving Quantum Chemistry Problems with First Generation Digital Quantum Computers. PhD thesis, ETH Zurich, 2021
work page 2021
-
[41]
Alan Bidart, Prateek Vaish, Tilas Kabengele, Yaoqi Pang, Yuan Liu, and Brenda M Rubenstein. Quantum computing beyond ground state electronic structure: A re- view of progress toward quantum chemistry out of the ground state.arXiv preprint arXiv:2509.19709, 2025
work page internal anchor Pith review arXiv 2025
-
[42]
Timothy N Georges, Marius Bothe, Christoph S¨ underhauf, Bjorn K Berntson, R´ obert Izs´ ak, and Aleksei V Ivanov. Quantum simulations of chemistry in first quantization with any basis set.npj Quantum Information, 11(1):55, 2025
work page 2025
-
[43]
Quan- tum algorithm for vibronic dynamics: case study on singlet fission solar cell design
Danial Motlagh, Robert A Lang, Paarth Jain, Jorge A Campos-Gonzalez-Angulo, William Maxwell, Tao Zeng, Alan Aspuru-Guzik, and Juan Miguel Arrazola. Quan- tum algorithm for vibronic dynamics: case study on singlet fission solar cell design. Quantum Science and Technology, 10(4):045048, 2025
work page 2025
-
[44]
Pauline J Ollitrault, Guglielmo Mazzola, and Ivano Tavernelli. Nonadiabatic molecular quantum dynamics with quantum computers.Physical Review Letters, 125(26):260511, 2020
work page 2020
-
[45]
Xiong Sun and William H Miller. Semiclassical initial value representation for electroni- cally nonadiabatic molecular dynamics.The Journal of chemical physics, 106(15):6346– 6353, 1997. 356
work page 1997
-
[46]
Dmitrii V Shalashilin and Mark S Child. Real time quantum propagation on a monte carlo trajectory guided grids of coupled coherent states: 26d simulation of pyrazine absorption spectrum.The Journal of chemical physics, 121(8):3563–3568, 2004
work page 2004
-
[47]
H-D Meyer, Uwe Manthe, and Lorenz S Cederbaum. The multi-configurational time- dependent hartree approach.Chemical Physics Letters, 165(1):73–78, 1990
work page 1990
-
[48]
Uwe Manthe, H-D Meyer, and Lorenz S Cederbaum. Wave-packet dynamics within the multiconfiguration Hartree framework: General aspects and application to NOCl. The Journal of chemical physics, 97(5):3199–3213, 1992
work page 1992
-
[49]
Haobin Wang. Multilayer multiconfiguration time-dependent hartree theory.The Jour- nal of Physical Chemistry A, 119(29):7951–7965, 2015
work page 2015
-
[50]
Oriol Vendrell and Hans-Dieter Meyer. Multilayer multiconfiguration time-dependent hartree method: Implementation and applications to a henon–heiles hamiltonian and to pyrazine.The Journal of Chemical Physics, 134(4), 2011
work page 2011
-
[51]
Density matrix formulation for quantum renormalization groups
Steven R White. Density matrix formulation for quantum renormalization groups. Physical review letters, 69(19):2863, 1992
work page 1992
-
[52]
Density-matrix algorithms for quantum renormalization groups.Phys- ical review b, 48(14):10345, 1993
Steven R White. Density-matrix algorithms for quantum renormalization groups.Phys- ical review b, 48(14):10345, 1993
work page 1993
-
[53]
The density-matrix renormalization group.Reviews of modern physics, 77(1):259–315, 2005
Ulrich Schollw¨ ock. The density-matrix renormalization group.Reviews of modern physics, 77(1):259–315, 2005
work page 2005
-
[54]
Ulrich Schollw¨ ock. The density-matrix renormalization group in the age of matrix product states.Annals of physics, 326(1):96–192, 2011
work page 2011
-
[55]
Time-evolution methods for matrix-product states
Sebastian Paeckel, Thomas K¨ ohler, Andreas Swoboda, Salvatore R Manmana, Ulrich Schollw¨ ock, and Claudius Hubig. Time-evolution methods for matrix-product states. Annals of Physics, 411:167998, 2019. 357
work page 2019
-
[56]
Michael Zwolak and Guifr´ e Vidal. Mixed-state dynamics in one-dimensional quantum lattice systems: A time-dependent superoperator renormalization algorithm.Physical review letters, 93(20):207205, 2004
work page 2004
-
[57]
Andrew John Daley, Corinna Kollath, Ulrich Schollw¨ ock, and Guifr´ e Vidal. Time- dependent density-matrix renormalization-group using adaptive effective hilbert spaces.Journal of Statistical Mechanics: Theory and Experiment, 2004(04):P04005, 2004
work page 2004
-
[58]
Jutho Haegeman, J Ignacio Cirac, Tobias J Osborne, Iztok Piˇ zorn, Henri Verschelde, and Frank Verstraete. Time-dependent variational principle for quantum lattices.Phys- ical review letters, 107(7):070601, 2011
work page 2011
-
[59]
Laurens Vanderstraeten, Jutho Haegeman, Philippe Corboz, and Frank Verstraete. Gradient methods for variational optimization of projected entangled-pair states.arXiv preprint arXiv:1606.09170, 2016
-
[60]
Pasquale Calabrese and John Cardy. Evolution of entanglement entropy in one- dimensional systems.Journal of Statistical Mechanics: Theory and Experiment, 2005(04):P04010, 2005
work page 2005
-
[61]
Cluster expansion analysis for delocalized systems
J ˇC´ ıˇ zek, J Paldus, and LˇSroubkov´ a. Cluster expansion analysis for delocalized systems. International Journal of Quantum Chemistry, 3(2):149–167, 1969
work page 1969
-
[62]
Coupled-cluster theory in quantum chemistry
Rodney J Bartlett and Monika Musia l. Coupled-cluster theory in quantum chemistry. Reviews of Modern Physics, 79(1):291–352, 2007
work page 2007
-
[63]
Henrik Koch, Ove Christiansen, Alfredo M Sanchez de Mer´ as, Trygve Helgaker, et al. The cc3 model: An iterative coupled cluster approach including connected triples.The Journal of chemical physics, 106(5):1808–1818, 1997. 358
work page 1997
-
[64]
Yannick J Bomble, John F Stanton, Mih´ aly K´ allay, and J¨ urgen Gauss. Coupled-cluster methods including noniterative corrections for quadruple excitations.The Journal of chemical physics, 123(5), 2005
work page 2005
-
[65]
Simen Kvaal, Hakon Richard Fredheim, Mads Greisen Højlund, and Thomas Bondo Pedersen. Time-dependent bivariational principle: Theoretical foundation for real-time propagation methods of coupled-cluster type.The Journal of Physical Chemistry A, 129(15):3508–3521, 2025
work page 2025
-
[66]
Andreas K¨ ohn and Attila Tajti. Can coupled-cluster theory treat conical intersections? The Journal of chemical physics, 127(4), 2007
work page 2007
-
[67]
Eirik F Kjønstad and Henrik Koch. An orbital invariant similarity constrained coupled cluster model.Journal of Chemical Theory and Computation, 15(10):5386–5397, 2019
work page 2019
-
[68]
Christian Huber and Tillmann Klamroth. Explicitly time-dependent coupled cluster singles doubles calculations of laser-driven many-electron dynamics.The Journal of chemical physics, 134(5), 2011
work page 2011
-
[69]
Anna I Krylov. Equation-of-motion coupled-cluster methods for open-shell and elec- tronically excited species: The hitchhiker’s guide to fock space.Annu. Rev. Phys. Chem., 59(1):433–462, 2008
work page 2008
-
[70]
Yannick J Bomble, Jamal C Saeh, John F Stanton, P´ eter G Szalay, Mih´ aly K´ allay, and J¨ urgen Gauss. Equation-of-motion coupled-cluster methods for ionized states with an approximate treatment of triple excitations.The Journal of chemical physics, 122(15), 2005
work page 2005
-
[71]
Ab initio quantum dynamics using coupled-cluster.The Journal of chemical physics, 136(19), 2012
Simen Kvaal. Ab initio quantum dynamics using coupled-cluster.The Journal of chemical physics, 136(19), 2012. 359
work page 2012
-
[72]
Takeshi Sato, Himadri Pathak, Yuki Orimo, and Kenichi L Ishikawa. Communication: Time-dependent optimized coupled-cluster method for multielectron dynamics.The Journal of chemical physics, 148(5), 2018
work page 2018
-
[73]
Himadri Pathak, Takeshi Sato, and Kenichi L Ishikawa. Time-dependent optimized coupled-cluster method with doubles and perturbative triples for first principles simu- lation of multielectron dynamics.Frontiers in Chemistry, 10:982120, 2022
work page 2022
-
[74]
Priyanka Pandey, Chen Qu, Apurba Nandi, Qi Yu, Paul L Houston, Riccardo Conte, and Joel M Bowman. Ab initio potential energy surface for NaCl–H2 with correct long-range behavior.The Journal of Physical Chemistry A, 128(5):902–908, 2024
work page 2024
-
[75]
Eirik F Kjønstad, Sara Angelico, and Henrik Koch. Coupled cluster theory for nonadia- batic dynamics: nuclear gradients and nonadiabatic couplings in similarity constrained coupled cluster theory.Journal of Chemical Theory and Computation, 20(16):7080– 7092, 2024
work page 2024
-
[76]
Rodney J Bartlett, Young Choon Park, Nicholas P Bauman, Ann Melnichuk, Duminda Ranasinghe, Moneesha Ravi, and Ajith Perera. Index of multi-determinantal and multi-reference character in coupled-cluster theory.The Journal of Chemical Physics, 153(23), 2020
work page 2020
-
[77]
H˚ akon Emil Kristiansen, Øyvind Sigmundson Schøyen, Simen Kvaal, and Thomas Bondo Pedersen. Numerical stability of time-dependent coupled-cluster meth- ods for many-electron dynamics in intense laser pulses.The Journal of chemical physics, 152(7), 2020
work page 2020
-
[78]
Eirik F Kjønstad and Henrik Koch. Biorthonormal formalism for nonadiabatic coupled cluster dynamics.Journal of Chemical Theory and Computation, 17(1):127–138, 2020
work page 2020
-
[79]
Time-dependent coupled-cluster formalism for the propagation of quantum superpositions
Martin A Mosquera. Time-dependent coupled-cluster formalism for the propagation of quantum superpositions. InSMT 2025. APS, 2025. 360
work page 2025
-
[80]
Benoit Mignolet and Basile FE Curchod. A walk through the approximations of ab initio multiple spawning.The Journal of chemical physics, 148(13), 2018
work page 2018
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.