A Data-Driven Parametric Reduced-Order Chemical Kinetics Model Derived from Atomistic Simulations
Pith reviewed 2026-05-21 00:10 UTC · model grok-4.3
The pith
A parametric autoencoder yields an interpretable reduced-order chemical kinetics model from atomistic simulations across temperatures.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The authors present a parametric, temperature-dependent autoencoder framework that learns a unified reduced-order description of chemical decomposition across a wide range of temperatures within a single model. Physical interpretability is enforced through non-negativity constraints and a softmax activation, enabling the latent variables to be directly associated with additive chemical components and their relative contributions. Reaction kinetics and heat-release parameters are optimized simultaneously within the neural-network architecture, providing a self-consistent coupling between chemical evolution and energetics. This yields significantly improved reconstruction accuracy compared to,
What carries the argument
Parametric temperature-dependent autoencoder with non-negativity constraints and softmax activation for learning additive chemical components.
If this is right
- Chemical evolution and energy release remain consistent because they are optimized together in the network.
- The model can describe decomposition at many temperatures using one set of parameters rather than separate models.
- Latent representations stay interpretable, allowing direct links to physical chemical species.
- Reconstruction of chemical data is more accurate than with conventional dimensionality reduction techniques.
Where Pith is reading between the lines
- Applying this framework to other materials or reaction types could extend its use beyond energetic materials to fields like catalysis or biochemistry.
- Validation against experimental measurements of reaction rates at various temperatures would strengthen confidence in the model's predictions.
- Embedding additional physical constraints, such as conservation laws, might enhance the model's reliability for long-term simulations.
Load-bearing premise
The non-negativity constraints and softmax activation will produce latent variables that correspond to real additive chemical components with optimizable kinetics and energetics.
What would settle it
A direct comparison showing whether the model's predicted species evolution and heat release match atomistic simulation results at a temperature outside the training set, or if the latent components fail to align with expected chemical species identities.
Figures







read the original abstract
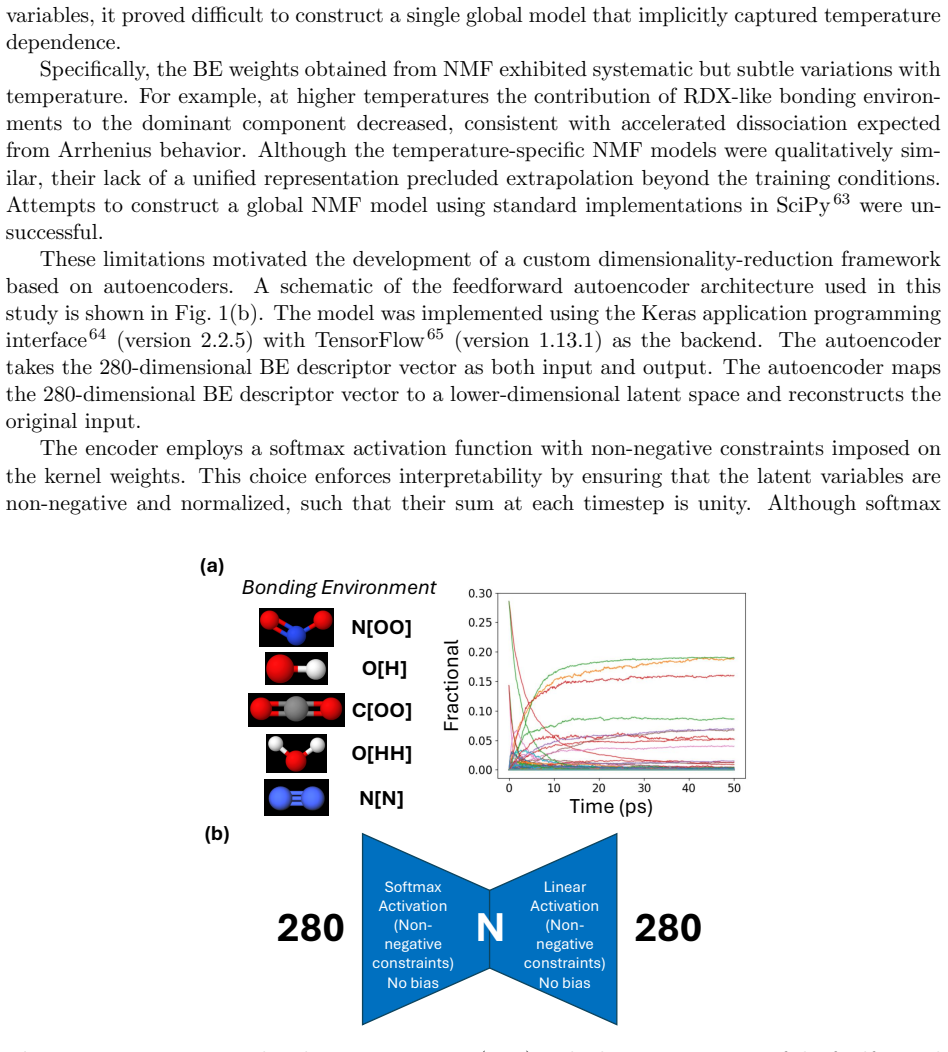
Coarse-grained modeling in molecular simulations serves not only to extend accessible time and length scales beyond atomistic limits, but also to reduce high-dimensional chemical data to low-dimensional representations that expose the underlying latent structure. In the context of energetic materials, reduced-order chemical kinetics models are essential for describing thermally driven decomposition, deflagration, and detonation. Recent data-driven approaches based on machine learning and dimensionality reduction have shown promise for constructing such models directly from atomistic simulations; however, when reaction pathways vary strongly with thermodynamic conditions, these methods can produce latent representations that are difficult to interpret physically or extrapolate reliably. Here, we introduce a parametric, temperature-dependent autoencoder framework that learns a unified reduced-order description of chemical decomposition across a wide range of temperatures within a single model. Physical interpretability is enforced through non-negativity constraints and a softmax activation, enabling the latent variables to be directly associated with additive chemical components and their relative contributions. Reaction kinetics and heat-release parameters are optimized simultaneously within the neural-network architecture, providing a self-consistent coupling between chemical evolution and energetics. The proposed approach yields significantly improved reconstruction accuracy compared to a state-of-the-art dimensionality-reduction method, as quantified by reductions in mean-squared error, while preserving a physically meaningful latent representation. These results demonstrate that parametric, interpretable machine-learning models can provide robust reduced-order chemical kinetics suitable for multiscale modeling of complex reactive systems.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces a parametric, temperature-dependent autoencoder that learns a unified reduced-order model of chemical decomposition from atomistic simulations of energetic materials. Non-negativity constraints and softmax activation are used to enforce that latent variables represent additive chemical components; reaction kinetics and heat-release parameters are optimized jointly inside the network. The central claim is that this yields lower mean-squared reconstruction error than a state-of-the-art dimensionality-reduction baseline while preserving a physically meaningful latent representation suitable for multiscale modeling.
Significance. If the interpretability and accuracy claims are substantiated, the method would supply a self-consistent, temperature-extrapolatable reduced-order kinetics model that couples chemistry and energetics, addressing a practical need in simulations of decomposition and detonation.
major comments (2)
- [§4.3 and Table 2] §4.3 and Table 2: the reported MSE reductions versus the baseline method are presented without error bars, without the exact baseline implementation details, and without a clear statement of the train/test split across temperatures; these omissions make it impossible to judge whether the improvement is statistically robust or merely an artifact of the particular data partitioning.
- [§3.2, Eq. (8)] §3.2, Eq. (8) and the accompanying text: the claim that non-negativity plus softmax produces latent variables that 'directly correspond to additive chemical components' is asserted from the architecture alone; no quantitative comparison to known species concentrations or reaction pathways from the underlying atomistic trajectories is shown, leaving open the possibility that the constraints are satisfied without recovering chemically meaningful decomposition channels.
minor comments (2)
- [Figure 3] Figure 3 caption: the temperature values used for the extrapolation test should be stated explicitly rather than referred to only as 'outside the training range.'
- [Methods] Notation: the symbol T_p for the parametric temperature input is introduced without a clear definition of its range or normalization; this should be added to the methods section.
Simulated Author's Rebuttal
We thank the referee for their careful reading of the manuscript and for the constructive comments. We address each of the major points below and indicate the revisions we will make to strengthen the presentation.
read point-by-point responses
-
Referee: [§4.3 and Table 2] §4.3 and Table 2: the reported MSE reductions versus the baseline method are presented without error bars, without the exact baseline implementation details, and without a clear statement of the train/test split across temperatures; these omissions make it impossible to judge whether the improvement is statistically robust or merely an artifact of the particular data partitioning.
Authors: We agree that the current presentation of the MSE results would benefit from additional statistical context. In the revised manuscript we will add error bars computed from multiple independent training runs with different random seeds, provide a detailed description of the baseline implementation (including any specific hyperparameters and software references), and explicitly state the train/test partitioning procedure used across the temperature range. These additions will allow readers to evaluate the robustness of the reported improvements. revision: yes
-
Referee: [§3.2, Eq. (8)] §3.2, Eq. (8) and the accompanying text: the claim that non-negativity plus softmax produces latent variables that 'directly correspond to additive chemical components' is asserted from the architecture alone; no quantitative comparison to known species concentrations or reaction pathways from the underlying atomistic trajectories is shown, leaving open the possibility that the constraints are satisfied without recovering chemically meaningful decomposition channels.
Authors: The non-negativity constraint together with the softmax activation mathematically guarantees that each latent vector consists of non-negative entries that sum to one, thereby representing additive fractional contributions by construction. This architectural choice is what enables the direct association with chemical components. While the manuscript does not contain a side-by-side quantitative match between the learned latent variables and specific species concentrations extracted from the atomistic trajectories, the physical utility of the representation is evidenced by the joint optimization with kinetics and heat-release terms and by the improved reconstruction fidelity. We will add a brief discussion of this point and, if feasible, a supplementary comparison in the revised version. revision: partial
Circularity Check
No significant circularity; derivation is self-contained
full rationale
The paper trains a parametric autoencoder on atomistic simulation data to produce a temperature-dependent reduced-order kinetics model. Reconstruction accuracy is measured via MSE against a baseline dimensionality-reduction method, and interpretability is imposed via non-negativity and softmax constraints within the network architecture. No equation or claim reduces the reported improvement or physical meaning to a fitted quantity defined by the same inputs, a self-citation chain, or a renaming of known results. The central results rest on empirical performance of the trained model rather than tautological re-expression of the training data.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Non-negativity constraints and softmax activation enforce that latent variables represent additive chemical components.
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel (J-cost uniqueness) unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
Physical interpretability is enforced through non-negativity constraints and a softmax activation, enabling the latent variables to be directly associated with additive chemical components... Reaction kinetics and heat-release parameters are optimized simultaneously
-
IndisputableMonolith/Foundation/RealityFromDistinction.leanreality_from_one_distinction unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
The encoded latent variables exhibit behavior consistent with both physical intuition... reactant-, intermediate-, and product-like components
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
Journal of Physics: Conference Series , volume=
Shock-induced hotspot formation and chemical reaction initiation in PETN containing a spherical void , author=. Journal of Physics: Conference Series , volume=. 2014 , organization=
work page 2014
-
[2]
The Journal of Physical Chemistry C , volume=
Ultrafast chemistry under nonequilibrium conditions and the shock to deflagration transition at the nanoscale , author=. The Journal of Physical Chemistry C , volume=. 2015 , publisher=
work page 2015
-
[3]
AIP conference proceedings , volume=
Ignition chemistry in HMX from thermal explosion to detonation , author=. AIP conference proceedings , volume=. 2002 , organization=
work page 2002
-
[4]
Combustion Theory and Modelling , volume=
Detonation waves in PBX 9501 , author=. Combustion Theory and Modelling , volume=. 2006 , publisher=
work page 2006
-
[5]
The Journal of Physical Chemistry , volume=
Critical conditions for impact-and shock-induced hot spots in solid explosives , author=. The Journal of Physical Chemistry , volume=. 1996 , publisher=
work page 1996
-
[6]
Combustion and Flame , volume=
Thermal decomposition models for HMX-based plastic bonded explosives , author=. Combustion and Flame , volume=. 2004 , publisher=
work page 2004
-
[7]
AIP Conference Proceedings , volume=
Modeling thermal ignition and the initial conditions for internal burning in PBX 9501 , author=. AIP Conference Proceedings , volume=. 2009 , organization=
work page 2009
-
[8]
Combustion and flame , volume=
Evaluation of reaction kinetics models for meso-scale simulations of hotspot initiation and growth in HMX , author=. Combustion and flame , volume=. 2020 , publisher=
work page 2020
-
[9]
Comparison of ReaxFF, DFTB, and DFT for phenolic pyrolysis. 2. Elementary reaction paths , author=. The Journal of Physical Chemistry A , volume=. 2013 , publisher=
work page 2013
-
[10]
Proceedings of the National Academy of Sciences , volume=
First-principles--based reaction kinetics from reactive molecular dynamics simulations: Application to hydrogen peroxide decomposition , author=. Proceedings of the National Academy of Sciences , volume=. 2019 , publisher=
work page 2019
-
[11]
Journal of chemical theory and computation , volume=
Mechanism of graphene formation via detonation synthesis: a DFTB nanoreactor approach , author=. Journal of chemical theory and computation , volume=. 2019 , publisher=
work page 2019
-
[12]
Combustion and Flame , volume=
Reactive molecular dynamics simulation and chemical kinetic modeling of pyrolysis and combustion of n-dodecane , author=. Combustion and Flame , volume=. 2011 , publisher=
work page 2011
-
[13]
Journal of chemical theory and computation , volume=
Automated discovery of reaction pathways, rate constants, and transition states using reactive molecular dynamics simulations , author=. Journal of chemical theory and computation , volume=. 2015 , publisher=
work page 2015
-
[14]
Journal of chemical information and modeling , volume=
Automated chemical kinetic modeling via hybrid reactive molecular dynamics and quantum chemistry simulations , author=. Journal of chemical information and modeling , volume=. 2018 , publisher=
work page 2018
-
[15]
Discovering chemistry with an ab initio nanoreactor , author=. Nature chemistry , volume=. 2014 , publisher=
work page 2014
-
[16]
Journal of chemical theory and computation , volume=
Automated discovery and refinement of reactive molecular dynamics pathways , author=. Journal of chemical theory and computation , volume=. 2016 , publisher=
work page 2016
-
[17]
The non-adiabatic nanoreactor: towards the automated discovery of photochemistry , author=. Chemical Science , volume=. 2021 , publisher=
work page 2021
-
[18]
The Journal of Physical Chemistry A , volume=
Nitromethane decomposition via automated reaction discovery and an ab initio corrected kinetic model , author=. The Journal of Physical Chemistry A , volume=. 2021 , publisher=
work page 2021
-
[19]
SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules , author=. Journal of chemical information and computer sciences , volume=. 1988 , publisher=
work page 1988
-
[20]
Development of CYP3A4 inhibition models: comparisons of machine-learning techniques and molecular descriptors , author=. SLAS Discovery , volume=. 2005 , publisher=
work page 2005
-
[21]
Chemometrics and Intelligent Laboratory Systems , volume=
Large-scale QSAR study of aromatase inhibitors using SMILES-based descriptors , author=. Chemometrics and Intelligent Laboratory Systems , volume=. 2014 , publisher=
work page 2014
-
[22]
Learning continuous and data-driven molecular descriptors by translating equivalent chemical representations , author=. Chemical science , volume=. 2019 , publisher=
work page 2019
-
[23]
Molecular Systems Design & Engineering , volume=
Deep learning for molecular design—a review of the state of the art , author=. Molecular Systems Design & Engineering , volume=. 2019 , publisher=
work page 2019
-
[24]
The Journal of chemical physics , volume=
Mirrored continuum and molecular scale simulations of the ignition of high-pressure phases of RDX , author=. The Journal of chemical physics , volume=. 2016 , publisher=
work page 2016
-
[25]
Combustion and Flame , volume=
Mirrored continuum and molecular scale simulations of deflagration in a nano-slab of HMX , author=. Combustion and Flame , volume=. 2020 , publisher=
work page 2020
-
[26]
Bulletin of the American Physical Society , year=
Unsupervised learning-based multiscale model of thermochemistry in 1, 3, 5-trinitro-1, 3, 5-triazine (RDX) , author=. Bulletin of the American Physical Society , year=
-
[27]
Propellants, Explosives, Pyrotechnics , volume=
Developing reaction chemistry models from reactive molecular dynamics: TATB , author=. Propellants, Explosives, Pyrotechnics , volume=. 2022 , publisher=
work page 2022
-
[28]
Estimation of heats of formation of organic compounds by additivity methods , author=. Chemical Reviews , volume=. 1993 , publisher=
work page 1993
-
[29]
New group contribution method for estimating properties of pure compounds , author=. AIChE Journal , volume=. 1994 , publisher=
work page 1994
-
[30]
The Journal of Physical Chemistry A , volume=
Atomic-level features for kinetic monte carlo models of complex chemistry from molecular dynamics simulations , author=. The Journal of Physical Chemistry A , volume=. 2021 , publisher=
work page 2021
-
[31]
The Journal of Chemical Physics , volume=
Heuristics for chemical species identification in dense systems , author=. The Journal of Chemical Physics , volume=. 2020 , publisher=
work page 2020
-
[32]
Exploring network structure, dynamics, and function using NetworkX , author=. 2007 , institution=
work page 2007
-
[33]
Journal of Chemical Theory and Computation , volume=
A quantum-based approach to predict primary radiation damage in polymeric networks , author=. Journal of Chemical Theory and Computation , volume=. 2020 , publisher=
work page 2020
-
[34]
Finding structure with randomness: Probabilistic algorithms for constructing approximate matrix decompositions , author=. SIAM review , volume=. 2011 , publisher=
work page 2011
-
[35]
Applied and Computational Harmonic Analysis , volume=
A randomized algorithm for the decomposition of matrices , author=. Applied and Computational Harmonic Analysis , volume=. 2011 , publisher=
work page 2011
-
[36]
Learning the parts of objects by non-negative matrix factorization , author=. nature , volume=. 1999 , publisher=
work page 1999
-
[37]
Proceedings of the 17th International Conference on Pattern Recognition, 2004
Application of non-negative and local non negative matrix factorization to facial expression recognition , author=. Proceedings of the 17th International Conference on Pattern Recognition, 2004. ICPR 2004. , volume=. 2004 , organization=
work page 2004
-
[38]
IEEE Transactions on Systems, Man, and Cybernetics, Part B (Cybernetics) , volume=
Graph-preserving sparse nonnegative matrix factorization with application to facial expression recognition , author=. IEEE Transactions on Systems, Man, and Cybernetics, Part B (Cybernetics) , volume=. 2010 , publisher=
work page 2010
-
[39]
Statistical Analysis and Data Mining: The ASA Data Science Journal , volume=
Nonnegative tensor decomposition with custom clustering for microphase separation of block copolymers , author=. Statistical Analysis and Data Mining: The ASA Data Science Journal , volume=. 2019 , publisher=
work page 2019
-
[40]
Machine Learning: Science and Technology , volume=
A neural network for determination of latent dimensionality in non-negative matrix factorization , author=. Machine Learning: Science and Technology , volume=. 2021 , publisher=
work page 2021
-
[41]
From Principal Subspaces to Principal Components with Linear Autoencoders
From principal subspaces to principal components with linear autoencoders , author=. arXiv preprint arXiv:1804.10253 , year=
work page internal anchor Pith review Pith/arXiv arXiv
-
[42]
Deep neural networks show an equivalent and often superior performance to dermatologists in onychomycosis diagnosis: Automatic construction of onychomycosis datasets by region-based convolutional deep neural network , author=. PloS one , volume=. 2018 , publisher=
work page 2018
-
[43]
European Journal of Cancer , volume=
Deep neural networks are superior to dermatologists in melanoma image classification , author=. European Journal of Cancer , volume=. 2019 , publisher=
work page 2019
-
[44]
arXiv preprint arXiv:2209.01862 , year=
Kinetics parameter optimization via neural ordinary differential equations , author=. arXiv preprint arXiv:2209.01862 , year=
-
[45]
The Journal of chemical physics , volume=
Algorithmic dimensionality reduction for molecular structure analysis , author=. The Journal of chemical physics , volume=. 2008 , publisher=
work page 2008
-
[46]
Dimensionality reduction methods for molecular simulations
Dimensionality reduction methods for molecular simulations , author=. arXiv preprint arXiv:1710.10629 , year=
work page internal anchor Pith review Pith/arXiv arXiv
-
[47]
The Journal of chemical physics , volume=
Collective variable discovery and enhanced sampling using autoencoders: Innovations in network architecture and error function design , author=. The Journal of chemical physics , volume=. 2018 , publisher=
work page 2018
-
[48]
Nature communications , volume=
Deep learning for universal linear embeddings of nonlinear dynamics , author=. Nature communications , volume=. 2018 , publisher=
work page 2018
-
[49]
The Journal of chemical physics , volume=
Time-lagged autoencoders: Deep learning of slow collective variables for molecular kinetics , author=. The Journal of chemical physics , volume=. 2018 , publisher=
work page 2018
-
[50]
Variational encoding of complex dynamics , author=. Physical Review E , volume=. 2018 , publisher=
work page 2018
-
[51]
Journal of chemical theory and computation , volume=
Transferable neural networks for enhanced sampling of protein dynamics , author=. Journal of chemical theory and computation , volume=. 2018 , publisher=
work page 2018
-
[52]
Stacked generalization , author=. Neural networks , volume=. 1992 , publisher=
work page 1992
-
[53]
Process modeling using stacked neural networks , author=. AIChE Journal , volume=. 1996 , publisher=
work page 1996
-
[54]
The Journal of Physical Chemistry A , volume=
Coupled thermal and electromagnetic induced decomposition in the molecular explosive HMX; a reactive molecular dynamics study , author=. The Journal of Physical Chemistry A , volume=. 2014 , publisher=
work page 2014
-
[55]
The Journal of Physical Chemistry C , volume=
Reactive molecular dynamics simulations to investigate the shock response of liquid nitromethane , author=. The Journal of Physical Chemistry C , volume=. 2019 , publisher=
work page 2019
-
[56]
The Journal of Physical Chemistry A , volume=
Insight into the chemistry of PETN under shock compression through ultrafast broadband mid-infrared absorption spectroscopy , author=. The Journal of Physical Chemistry A , volume=. 2020 , publisher=
work page 2020
-
[57]
Parallel reactive molecular dynamics: Numerical methods and algorithmic techniques , author=. parallel computing , volume=. 2012 , publisher=
work page 2012
-
[58]
Journal of computational physics , volume=
Fast parallel algorithms for short-range molecular dynamics , author=. Journal of computational physics , volume=. 1995 , publisher=
work page 1995
-
[59]
The Journal of Physical Chemistry C , volume=
Role of Molecular Disorder on the Reactivity of RDX , author=. The Journal of Physical Chemistry C , volume=. 2018 , publisher=
work page 2018
-
[60]
Redetermination of cyclo-trimethylenetrinitramine , author=. Structure Reports , volume=. 2008 , publisher=
work page 2008
-
[61]
IEICE transactions on fundamentals of electronics, communications and computer sciences , volume=
Fast local algorithms for large scale nonnegative matrix and tensor factorizations , author=. IEICE transactions on fundamentals of electronics, communications and computer sciences , volume=. 2009 , publisher=
work page 2009
-
[62]
Algorithms for nonnegative matrix factorization with the -divergence , author=. Neural computation , volume=. 2011 , publisher=
work page 2011
-
[63]
SciPy 1.0: fundamental algorithms for scientific computing in Python , author=. Nature methods , volume=. 2020 , publisher=
work page 2020
-
[64]
Proceedings of the IEEE conference on computer vision and pattern recognition , pages=
Xception: Deep learning with depthwise separable convolutions , author=. Proceedings of the IEEE conference on computer vision and pattern recognition , pages=
-
[65]
TensorFlow: Large-Scale Machine Learning on Heterogeneous Distributed Systems
Tensorflow: Large-scale machine learning on heterogeneous distributed systems , author=. arXiv preprint arXiv:1603.04467 , year=
work page internal anchor Pith review Pith/arXiv arXiv
-
[66]
A Critical Review of Recurrent Neural Networks for Sequence Learning
A critical review of recurrent neural networks for sequence learning , author=. arXiv preprint arXiv:1506.00019 , year=
work page internal anchor Pith review Pith/arXiv arXiv
-
[67]
Learning Adversary-Resistant Deep Neural Networks
Learning adversary-resistant deep neural networks , author=. arXiv preprint arXiv:1612.01401 , year=
work page internal anchor Pith review Pith/arXiv arXiv
-
[68]
Strang, in Linear Algebra and Its Applications, 2nd ed
G. Strang, in Linear Algebra and Its Applications, 2nd ed. (Academic Press, Inc., Orlando, FL, 1980), pp. 139--142
work page 1980
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.