Approximate Numerical Integration of the Chemical Master Equation for Stochastic Reaction Networks
Pith reviewed 2026-05-24 16:45 UTC · model grok-4.3
The pith
A dynamical state space truncation procedure paired with adaptive ODE solvers approximates solutions to the chemical master equation for large stochastic reaction networks.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
We present an approximate numerical integration approach that combines a dynamical state space truncation procedure with efficient numerical integration schemes for systems of ordinary differential equations including adaptive step size selection based on local error estimates. The efficiency and accuracy is demonstrated by numerical examples.
What carries the argument
dynamical state space truncation procedure combined with adaptive-step-size ODE integration
If this is right
- The method handles state spaces that are huge or formally infinite by keeping only a dynamically adjusted finite subset of states.
- Stiffness arising from widely separated time scales is managed through adaptive step-size selection that respects local error bounds.
- Numerical examples confirm that the combined truncation-plus-integration scheme runs faster than full-state methods while retaining acceptable accuracy.
- The resulting probability distributions can be used to compute moments or other statistics of the stochastic process without solving the full infinite system.
Where Pith is reading between the lines
- The same truncation-plus-adaptive-integration pattern could be tested on other continuous-time Markov chains outside biochemistry, such as queueing networks.
- If truncation thresholds are made state-dependent in a more refined way, the method might extend to networks whose probability mass spreads over time rather than remaining localized.
- One could measure the computational gain by comparing run times against fixed-state-space solvers on networks with known scaling behavior.
Load-bearing premise
The dynamical state space truncation procedure can be performed without losing critical probability mass or introducing uncontrolled errors for the reaction networks under consideration.
What would settle it
Apply the method to a small, exactly solvable reaction network, compute the full probability distribution at several time points, and check whether the truncated approximation deviates by more than a chosen tolerance from the exact solution.
Figures
read the original abstract
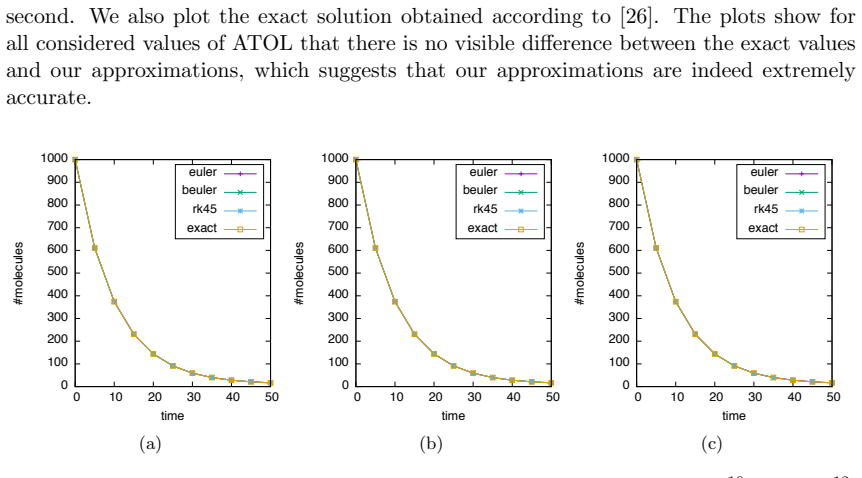
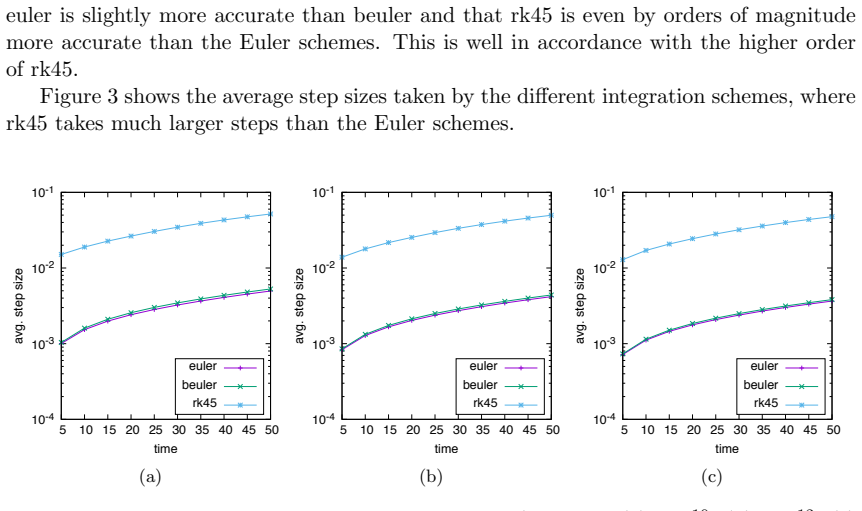
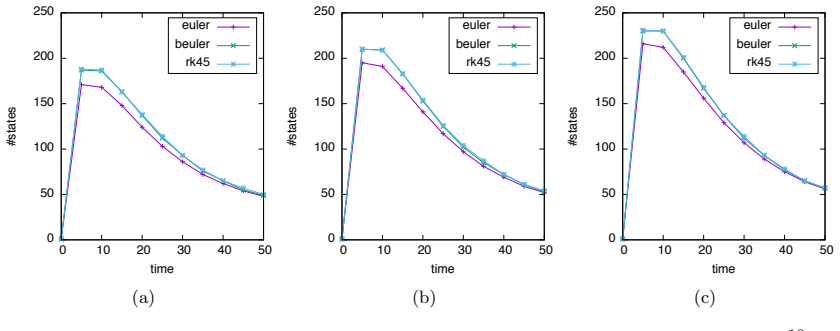
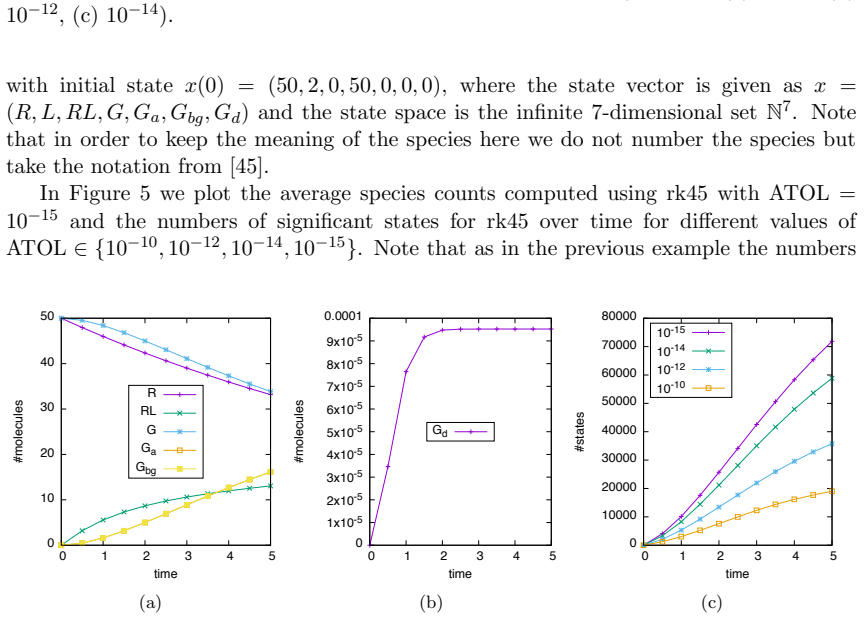
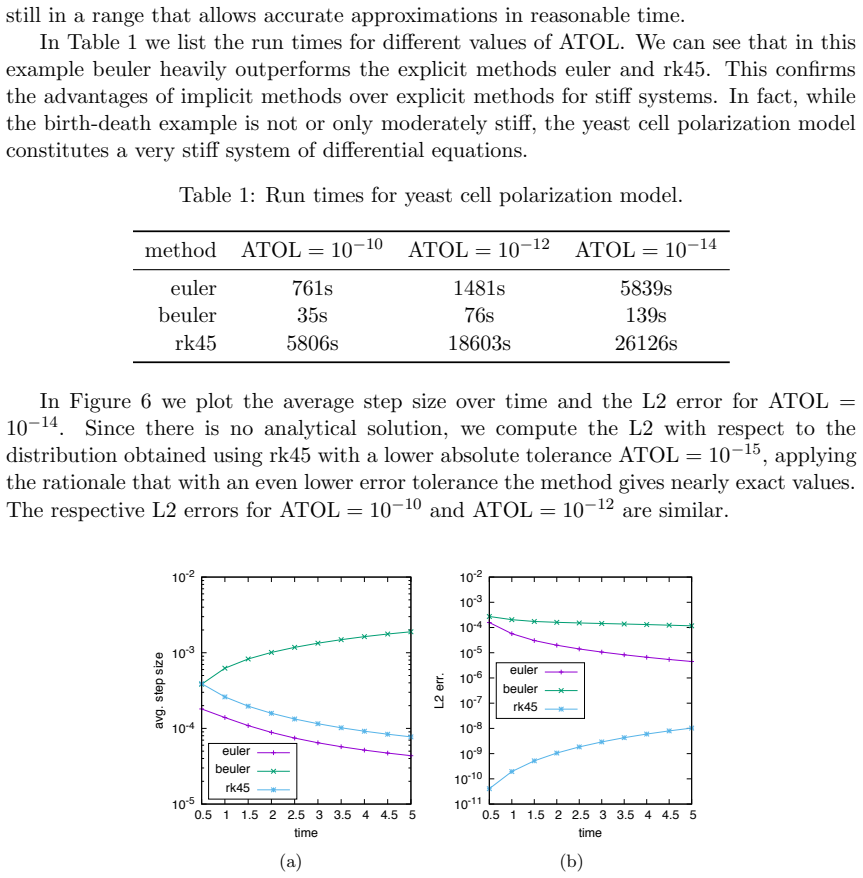
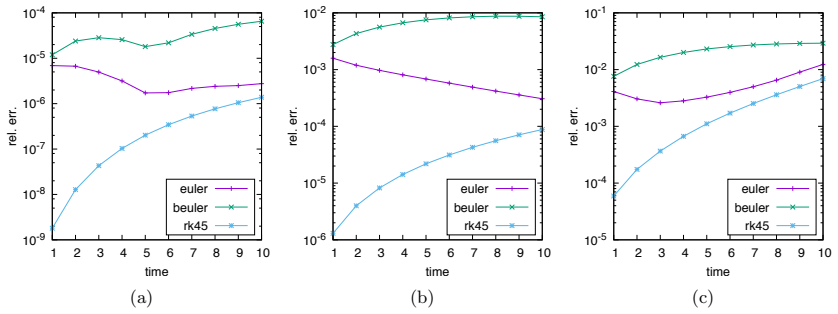
Numerical solution of the chemical master equation for stochastic reaction networks typically suffers from the state space explosion problem due to the curse of dimensionality and from stiffness due to multiple time scales. The dimension of the state space equals the number of molecular species involved in the reaction network and the size of the system of differential equations equals the number of states in the corresponding continuous-time Markov chain, which is usually enormously huge and often even infinite. Thus, efficient numerical solution approaches must be able to handle huge, possibly infinite and stiff systems of differential equations efficiently. We present an approximate numerical integration approach that combines a dynamical state space truncation procedure with efficient numerical integration schemes for systems of ordinary differential equations including adaptive step size selection based on local error estimates. The efficiency and accuracy is demonstrated by numerical examples.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes an approximate numerical method for solving the chemical master equation (CME) of stochastic reaction networks. It combines a dynamical state-space truncation procedure with adaptive ODE integrators (including local error-based step-size control) to mitigate the curse of dimensionality and stiffness. Efficiency and accuracy are asserted on the basis of numerical examples.
Significance. If the truncation step can be equipped with explicit probability-mass bounds and interaction analysis with stiffness, the method would supply a practical computational tool for large reaction networks. The adaptive ODE component is standard and correctly applied, but the absence of any a-priori error control on truncation reduces the work to an empirical illustration rather than a general, verifiable algorithm.
major comments (3)
- [Abstract] Abstract: the headline claim of an 'approximate numerical integration approach' that is both efficient and accurate rests on the dynamical truncation step, yet the abstract supplies neither the truncation rule, a tail-probability bound, nor any statement of how truncation error interacts with the adaptive integrator.
- [Method (truncation)] Method description (truncation procedure): no a-priori bound is given on the probability mass discarded at each truncation step, nor is there analysis of bias introduced into the retained dynamics when the network is stiff or when rare-event probabilities matter.
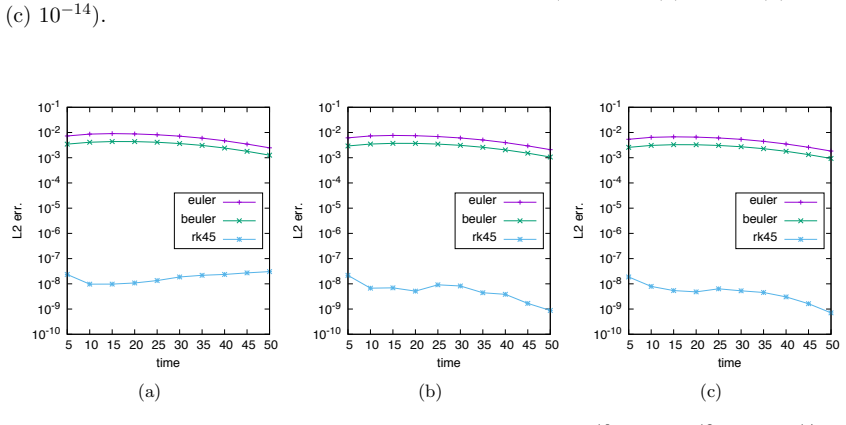
- [Numerical examples] Numerical examples: the examples demonstrate behavior on chosen instances but contain no quantitative comparison against exact solutions (where feasible) or against established truncation methods with known error guarantees, so they cannot substantiate the general claim.
minor comments (1)
- [Method] Notation for the truncated state space and the projection operator should be introduced once and used consistently; currently the same symbol appears to be overloaded.
Simulated Author's Rebuttal
We thank the referee for the detailed and constructive report. We address each major comment below, indicating planned revisions to strengthen the manuscript while noting where the work remains empirical in nature.
read point-by-point responses
-
Referee: [Abstract] Abstract: the headline claim of an 'approximate numerical integration approach' that is both efficient and accurate rests on the dynamical truncation step, yet the abstract supplies neither the truncation rule, a tail-probability bound, nor any statement of how truncation error interacts with the adaptive integrator.
Authors: We agree the abstract is too concise. We will revise it to briefly outline the dynamical truncation rule (state-space reduction based on probability mass thresholds) and clarify that truncation error is controlled empirically through the adaptive ODE integrator's local error estimates rather than via a priori tail bounds. revision: yes
-
Referee: [Method (truncation)] Method description (truncation procedure): no a-priori bound is given on the probability mass discarded at each truncation step, nor is there analysis of bias introduced into the retained dynamics when the network is stiff or when rare-event probabilities matter.
Authors: The method is presented as an approximate, adaptive procedure without general a-priori bounds, as deriving such bounds for arbitrary stiff networks is nontrivial and not claimed. We will add a dedicated discussion subsection acknowledging the lack of explicit probability-mass guarantees, noting the potential for bias in rare-event regimes, and recommending that practitioners monitor discarded mass at runtime. The adaptive integrator mitigates stiffness on the retained states but does not eliminate truncation bias. revision: partial
-
Referee: [Numerical examples] Numerical examples: the examples demonstrate behavior on chosen instances but contain no quantitative comparison against exact solutions (where feasible) or against established truncation methods with known error guarantees, so they cannot substantiate the general claim.
Authors: We will expand the numerical section to include, for all small networks where exact solutions are computationally feasible, direct quantitative comparisons (e.g., total variation distance or moment errors) against the exact CME solution obtained by matrix exponentiation. Where applicable, we will also add comparisons to the finite-state projection method to provide context against an established truncation approach. revision: yes
Circularity Check
No circularity; derivation is a standard procedural numerical method
full rationale
The paper describes a combination of dynamical state-space truncation with adaptive ODE integrators for approximating solutions to the chemical master equation. No equations, parameters, or claims reduce to self-definitions, fitted inputs renamed as predictions, or load-bearing self-citations. The approach is presented as an algorithmic procedure whose accuracy is illustrated by examples rather than derived from prior results by the same authors. The central claim remains independent of its inputs and does not rely on any of the enumerated circularity patterns.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
D. F. Anderson. Incorporating postleap checks in tau-leaping. Journal of Chemical Physics, 128(5):054103, 2008
work page 2008
-
[2]
A. Andreychenko, W. Sandmann, and V. Wolf. Approximate adative uniformization of continuous-time Markov chains.Applied Mathematical Modelling, 61:561–576, 2018
work page 2018
-
[3]
A. B. Bortz, M. H. Kalos, and J. L. Lebowitz. A new algorithm for Monte Carlo simulation of Ising spin systems. Journal of Computational Physics , 17:10–18, 1975. 18
work page 1975
-
[4]
K. Burrage, M. Hegland, S. MacNamara, and R. Sidje. A Krylov-based finite state projection algorithm for solving the chemical master equation arising in the discrete modelling of biological systems. In A. N. Langville and W. J. Stewart, editors, Pro- ceedings of the Markov Anniversary Meeting: An International Conference to Cele- brate the 150th Anniversar...
work page 2006
-
[5]
K. Burrage, T. Tian, and P. Burrage. A multi-scaled approach for simulating chemical reaction systems. Progress in Biophysics and Molecular Biology , 85:217–234, 2004
work page 2004
-
[6]
H. Busch, W. Sandmann, and V. Wolf. A numerical aggregation algorithm for the enzyme-catalyzed substrate conversion. In Proceedings of the 2006 International Con- ference on Computational Methods in Systems Biology , volume 4210 of Lecture Notes in Computer Science , pages 298–311. Springer, 2006
work page 2006
-
[7]
J. C. Butcher. Numerical Methods for Ordinary Differential Equations . John Wiley & Sons, 2nd edition, 2008
work page 2008
-
[8]
Y. Cao, D. T. Gillespie, and L. R. Petzold. Efficient stepsize selection for the tau- leaping simulation. Journal of Chemical Physics , 124:044109–144109–11, 2006
work page 2006
-
[9]
Y. Cao, D. T. Gillespie, and L. R. Petzold. The adaptive explicit-implicit tau-leaping method with automatic tau selection. Journal of Chemical Physics , 126(22):224101, 2007
work page 2007
-
[10]
Y. Cao, H. Li, and L. R. Petzold. Efficient formulation of the stochastic simulation algorithm for chemically reacting systems. Journal of Chemical Physics, 121(9):4059– 4067, 2004
work page 2004
-
[11]
C.-S. Chou, Q. Nie, and T.-M. Yi. Modeling robustness tradeoffs in yeast cell polar- ization induced by spatial gradients. PLoS ONE, 3(9):e3103, 2008
work page 2008
-
[12]
E. de Souza e Silva and P. M. Ochoa. State space exploration in Markov models. In Proceedings of the ACM SIGMETRICS Joint International Conference on Measure- ment and Modeling of Computer Systems , pages 152–166, 1992
work page 1992
-
[13]
J. R. Dormand and P. J. Prince. A family of embedded Runge-Kutta formulae. Journal of Computational and Applied Mathematics , 6(1):19–26, 1980
work page 1980
-
[14]
W. E., D. Liu, and E. Vanden-Eijnden. Nested stochastic simulation algorithms for chemical kinetic systems with multiple time scales.Journal of Computational Physics, 221:158–180, 2007
work page 2007
-
[15]
M. A. Gibson and J. Bruck. Efficient exact stochastic simulation of chemical systems with many species and many channels. Journal of Physical Chemistry , A 104:1876– 1889, 2000
work page 2000
-
[16]
D. T. Gillespie. A general method for numerically simulating the time evolution of coupled chemical reactions. Journal of Computational Physics , 22:403–434, 1976. 19
work page 1976
-
[17]
D. T. Gillespie. Exact stochastic simulation of coupled chemical reactions. Journal of Physical Chemistry , 71(25):2340–2361, 1977
work page 1977
-
[18]
D. T. Gillespie. A rigorous derivation of the chemical master equation. Physica A, 188:404–425, 1992
work page 1992
-
[19]
D. T. Gillespie. Approximate accelerated stochastic simulation of chemically reacting systems. Journal of Chemical Physics , 115:1716–1732, 2001
work page 2001
- [20]
-
[21]
E. Hairer and G. Wanner. Solving Ordinary Differential Equations II: Stiff and Differential-Algebraic Problems. Springer-Verlag, Berlin Heidelberg, 2nd edition, 1996
work page 1996
-
[22]
L. A. Harris and P. Clancy. A partitioned leaping approach for multiscale modeling of chemical reaction dynamics. Journal of Chemical Physics , 125(14):144107, 2006
work page 2006
-
[23]
E. L. Haseltine and J. B. Rawlings. Approximate simulation of coupled fast and slow reactions for stochastic chemical kinetics. Journal of Chemical Physics, 117(15):6959– 6969, 2002
work page 2002
-
[24]
A. Hellander and P. L¨ otstedt. Hybrid method for the chemical master equation. Journal of Computational Physics , 227:100–122, 2007
work page 2007
-
[25]
T. Jahnke. An adaptive wavelet method for the chemical master equation. SIAM Journal on Scientific Computing , 31(6):4373–4394, 2010
work page 2010
-
[26]
T. Jahnke and W. Huisinga. Solving the chemical master equation for monomolecular reactions analytically. Journal of Mathematical Biology , 54(1):1–26, 2007
work page 2007
-
[27]
T. Jahnke and T. Udrescu. Solving chemical master equations by adaptive wavelet compression. Journal of Computational Physics , 229:5724–5741, 2010
work page 2010
-
[28]
T. G. Kurtz. The relationship between stochastic and deterministic models for chem- ical reactions. Journal of Chemical Physics , 57(7):2976–2978, 1972
work page 1972
-
[29]
I. J. Laurenzi. An analytical solution of the stochastic master equation for reversible bimolecular reaction kinetics. Journal of Chemical Physics , 113(8):3315–3322, 2000
work page 2000
- [30]
-
[31]
S. MacNamara, A. M. Bersani, K. Burrage, and R. B. Sidje. Stochastic chemical kinetics and the total quasi-steady-state assumption: application to the stochastic simulation algorithm and chemical master equation. Journal of Chemical Physics , 129:095105, 2008. 20
work page 2008
-
[32]
S. MacNamara, K. Burrage, and R. B. Sidje. Multiscale modeling of chemical kinetics via the master equation. Multiscale Modeling & Simulation , 6(4):1146–1168, 2008
work page 2008
-
[33]
M. Mateescu, V. Wolf, F. Didier, and T.A. Henzinger. Fast adaptive uniformisation of the chemical master equation. IET Systems Biology , 4(6):441–452, 2010
work page 2010
-
[34]
J. M. McCollum, G. D. Peterson, C. D. Cox, M. L. Simpson, and N. F. Samatova. The sorting direct method for stochastic simulation of biochemical systems with varying reaction execution behavior. Computational Biology and Chemistry , 30(1):39–49, 2006
work page 2006
- [35]
-
[36]
L. Mikeev, W. Sandmann, and V. Wolf. Numerical approximation of rare event probabilities in biochemically reacting systems. In A. Gupta and T. A. Henzinger, editors, Proceedings of the 11th International Conference on Computational Methods in Systems Biology, CMSB , volume 8130 of Lecture Notes in Bioinformatics , pages 5–18. Springer, 2013
work page 2013
-
[37]
C. Moler and C. Van Loan. Nineteen dubious ways to compute the exponential of a matrix. SIAM Review, 20:801–836, 1978
work page 1978
-
[38]
C. Moler and C. Van Loan. Nineteen dubious ways to compute the exponential of a matrix, twenty-five years later. SIAM Review, 45:3–49, 2003
work page 2003
-
[39]
B. Munsky and M. Khammash. The finite state projection algorithm for the solution of the chemical master equation. Journal of Chemical Physics , 124:044104, 2006
work page 2006
-
[40]
B. Munsky and M. Khammash. A multiple time interval finite state projection al- gorithm for the solution to the chemical master equation. Journal of Computational Physics, 226:818–835, 2007
work page 2007
-
[41]
D. Pruyne and A. Bretcher. Polarization of cell growth in yeast I. Establishment and maintenance of polarity states. Journal of Cell Science , 113:365–375, 2000
work page 2000
-
[42]
C. V. Rao and A. Arkin. Stochastic chemical kinetics and the quasi-steady-state- assumption: Application to the Gillespie algorithm. Journal of Chemical Physics , 118:4999–5010, 2003
work page 2003
-
[43]
M. Rathinam and H. El Samad. Reversible-equivalent-monomolecular tau: A leaping method for small number and stiff stochastic chemical systems. Journal of Compu- tational Physics, 224:897–923, 2007
work page 2007
-
[44]
M. Rathinam, L. R. Petzold, Y. Cao, and D. T. Gillespie. Stiffness in stochastic chemically reacting systems: The implicit tau-leaping method. Journal of Chemical Physics, 119:12784–12794, 2003. 21
work page 2003
-
[45]
M. K. Roh, B. J. Daigle Jr., D. T. Gillespie, and L. R. Petzold. State-dependent doubly weighted stochastic simulation algorithm for automatic characterization of stochastic biochemical rare events. Journal of Chemical Physics , 135:234108, 2011
work page 2011
- [46]
-
[47]
H. Salis and Y. Kaznessis. Accurate hybrid stochastic simulation of a system of coupled chemical or biochemical reactions. Journal of Chemical Physics , 122:054103, 2005
work page 2005
- [48]
- [49]
- [50]
-
[51]
W. Sandmann and V. Wolf. Computational probability for systems biology. In Pro- ceedings of the Workshop on Formal Methods in Systems Biology , volume 5054 of Lecture Notes in Computer Science , pages 33–47. Springer, 2008
work page 2008
-
[52]
L. F. Shampine, I. Gladwell, and S. Thompson. Solving ODEs with MATLAB . Cam- bridge University Press, 2003
work page 2003
-
[53]
T. Tian and K. Burrage. Binomial leap methods for simulating stochastic chemical kinetics. Journal of Chemical Physics , 121:10356–10364, 2004
work page 2004
-
[54]
N. van Kampen. Stochastic Processes in Physics and Chemistry . Elsevier, North- Holland, 1992
work page 1992
-
[55]
K. Vasudeva and U. S. Bhalla. Adaptive stochastic-deterministic chemical kinetics simulation. Bioinformatics, 20(1):78–84, 2004
work page 2004
- [56]
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.