Detecting Evolutionary Change-Points with Branch-Specific Substitution Models and Shrinkage Priors
Pith reviewed 2026-05-19 05:15 UTC · model grok-4.3
The pith
Combining branch-specific substitution models with shrinkage priors allows automatic identification of evolutionary change-points without prior knowledge of their locations.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
By integrating branch-specific substitution models with shrinkage priors, it is possible to automatically identify change-points in evolutionary dynamics on a phylogeny while simultaneously estimating distinct substitution parameters for each branch, enabled by a new analytical gradient algorithm whose computational time scales linearly with the number of parameters.
What carries the argument
Shrinkage priors on the branch-specific substitution parameters that automatically identify change-points by shrinking non-change branches to shared values, paired with an analytical gradient for efficient optimization.
Load-bearing premise
The shrinkage priors correctly distinguish true evolutionary change-points from random statistical noise in the data without missing real shifts or creating false ones.
What would settle it
A simulation study where known change-points are inserted into sequence data and the method fails to recover them accurately or the analytical gradient produces likelihood values differing from numerical checks.
Figures



read the original abstract
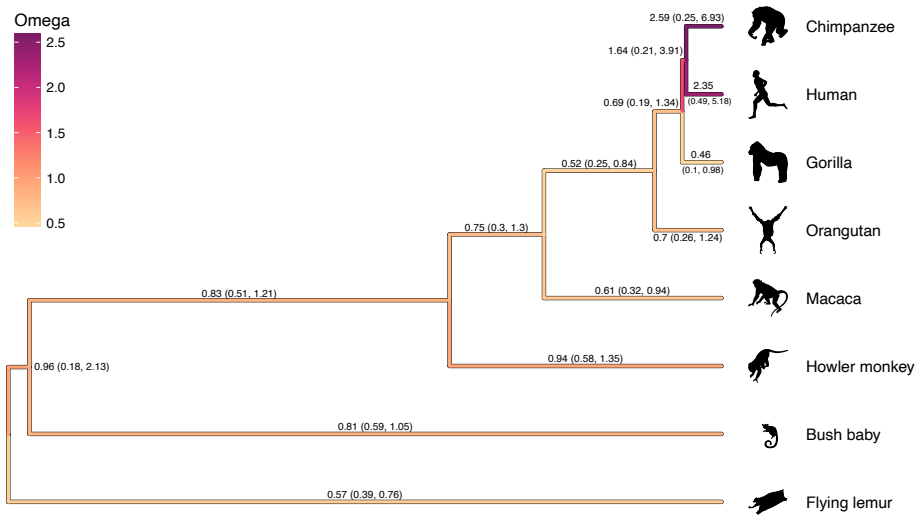
Branch-specific substitution models are popular for detecting evolutionary change-points, such as shifts in selective pressure. However, applying such models typically requires prior knowledge of change-point locations on the phylogeny or faces scalability issues with large data sets. To address both limitations, we integrate branch-specific substitution models with shrinkage priors to automatically identify change-points without prior knowledge, while simultaneously estimating distinct substitution parameters for each branch. To enable tractable inference under this high-dimensional model, we develop an analytical gradient algorithm for the branch-specific substitution parameters where the computational time is linear in the number of parameters. We apply this gradient algorithm to infer selection pressure dynamics in the evolution of the BRCA1 gene in primates and mutational dynamics in viral sequences from the recent mpox epidemic. Our novel algorithm enhances inference efficiency, achieving up to a 126-fold speedup per iteration in maximum likelihood optimization when compared to central difference numerical gradient method and up to a 2026-fold improvement in computational performance within a Bayesian framework using Hamiltonian Monte Carlo sampler compared to conventional univariate random walk sampler.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes integrating branch-specific substitution models with shrinkage priors to enable automatic detection of evolutionary change-points on phylogenies without prior specification of their locations. It develops an analytical gradient algorithm for the branch-specific parameters whose per-iteration cost is stated to be linear in the number of parameters, and demonstrates the approach on BRCA1 selection dynamics in primates and mutational dynamics in mpox viral sequences, reporting up to 126-fold speedup versus central-difference gradients in ML optimization and up to 2026-fold improvement versus univariate random-walk sampling in HMC.
Significance. If the gradient derivation is exact and the shrinkage priors recover change-points with controlled bias and power, the method would address a practical scalability barrier in high-dimensional phylogenetic models and facilitate routine inference of selection or rate shifts on larger trees.
major comments (2)
- [Abstract and Results] Abstract and Results sections: the central claim of reliable automatic change-point detection is not supported by any reported quantitative validation (simulation recovery rates, false-positive rates under known shifts, or cross-validation performance). Only computational timings are supplied; this is load-bearing for the claim that the shrinkage prior correctly separates signal from noise.
- [Methods, analytical gradient derivation] Methods, analytical gradient derivation: the asserted linear scaling in the number of branch-specific parameters does not explicitly account for the additional tree traversals or partial-likelihood storage required once the Felsenstein pruning algorithm is applied to a fully branch-specific model; the shrinkage prior further couples all parameters, potentially introducing overhead not captured by the single-pass assumption.
minor comments (2)
- [Methods] The manuscript should state the precise form of the shrinkage prior (e.g., Laplace, horseshoe) and how its hyperparameters are set or inferred, since these are the only free parameters listed.
- [Figures and Tables] Figure legends and table captions should explicitly indicate whether reported speedups are wall-clock times, iteration counts, or effective sample sizes per unit time.
Simulated Author's Rebuttal
We thank the referee for their constructive comments, which help clarify the scope and presentation of our work. We address each major comment below.
read point-by-point responses
-
Referee: [Abstract and Results] Abstract and Results sections: the central claim of reliable automatic change-point detection is not supported by any reported quantitative validation (simulation recovery rates, false-positive rates under known shifts, or cross-validation performance). Only computational timings are supplied; this is load-bearing for the claim that the shrinkage prior correctly separates signal from noise.
Authors: We agree that the manuscript would be strengthened by quantitative validation of change-point recovery. The current results focus on real-data applications and computational performance, but we will add a dedicated simulation study reporting recovery rates, false-positive rates, and power under known shift scenarios. This addition will be included in the revised version. revision: yes
-
Referee: [Methods, analytical gradient derivation] Methods, analytical gradient derivation: the asserted linear scaling in the number of branch-specific parameters does not explicitly account for the additional tree traversals or partial-likelihood storage required once the Felsenstein pruning algorithm is applied to a fully branch-specific model; the shrinkage prior further couples all parameters, potentially introducing overhead not captured by the single-pass assumption.
Authors: The gradient algorithm performs a single forward-backward traversal to obtain all partial likelihoods and their derivatives simultaneously, so the dominant cost remains linear in the number of branch-specific parameters even under a fully branch-specific model. The shrinkage prior gradient is computed in an additional linear pass that does not require extra traversals. We will revise the Methods section to make these steps and the resulting complexity explicit. revision: partial
Circularity Check
No significant circularity; algorithmic construction is independent
full rationale
The paper develops a new analytical gradient for branch-specific substitution parameters and pairs it with shrinkage priors for change-point detection. Performance is benchmarked against external baselines (central-difference numerical gradients and univariate random-walk samplers) rather than being defined in terms of its own fitted outputs. No load-bearing step reduces by construction to a self-citation, a fitted parameter renamed as prediction, or an ansatz smuggled via prior work. The derivation chain is self-contained against external benchmarks and does not exhibit the enumerated circularity patterns.
Axiom & Free-Parameter Ledger
free parameters (1)
- shrinkage prior hyperparameters
axioms (1)
- domain assumption The underlying continuous-time Markov substitution process on each branch is correctly specified by the chosen model family.
Lean theorems connected to this paper
-
IndisputableMonolith/Foundation/AbsoluteFloorClosure.leanreality_from_one_distinction unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
We develop an analytical gradient algorithm for the branch-specific substitution parameters where the computational time is linear in the number of parameters... using the post- and pre-order partial likelihood vectors... spectral representations approach to calculate the first directional derivative of the matrix exponential
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
we employ shrinkage priors on ϕi’s... Bayesian bridge prior... to shrink the total number of substitution parameter changes along the tree
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
Al-Mohy, A. H. and Higham, N. J. 2011. Computing the action of the matrix exponential, with an application to exponential integrators. SIAM journal on scientific computing , 33(2): 488–511. ´Alvarez-Carretero, S., Kapli, P., and Yang, Z. 2023. Beginner’s guide on the use of paml to detect positive selection. Molecular biology and evolution , 40(4): msad041
work page 2011
-
[2]
Ayres, D. L., Cummings, M. P., Baele, G., Darling, A. E., Lewis, P. O., Swofford, D. L., Huelsen- beck, J. P., Lemey, P., Rambaut, A., and Suchard, M. A. 2019. BEAGLE 3: Improved perfor- mance, scaling, and usability for a high-performance computing library for statistical phyloge- netics. Syst Biol., 68(6): 1052–1061
work page 2019
-
[3]
Baele, G., Lemey, P., Bedford, T., Rambaut, A., Suchard, M. A., and Alekseyenko, A. V. 2012. Improving the accuracy of demographic and molecular clock model comparison while accommo- dating phylogenetic uncertainty. Molecular biology and evolution , 29(9): 2157–2167
work page 2012
-
[4]
A., Bielejec, F., and Lemey, P
Baele, G., Suchard, M. A., Bielejec, F., and Lemey, P. 2016. Bayesian codon substitution modelling to identify sources of pathogen evolutionary rate variation. Microbial Genomics , 2(6): e000057
work page 2016
-
[5]
S., Bastide, P., Lemey, P., and Suchard, M
Baele, G., Gill, M. S., Bastide, P., Lemey, P., and Suchard, M. A. 2021. Markov-modulated continuous-time markov chains to identify site-and branch-specific evolutionary variation in beast. Systematic biology , 70(1): 181–189
work page 2021
-
[6]
J., Rambaut, A., and Suchard, M
Drummond, A. J., Rambaut, A., and Suchard, M. A. 2025. BEAST X for Bayesian phylogenetic, phylogeographic and phylodynamic inference. Nature Methods
work page 2025
-
[7]
Boussau, B. and Gouy, M. 2006. Efficient likelihood computations with nonreversible models of evolution. Systematic biology , 55(5): 756–768
work page 2006
-
[8]
Carvalho, C. M., Polson, N. G., and Scott, J. G. 2010. The horseshoe estimator for sparse signals. Biometrika, 97(2): 465–480
work page 2010
-
[9]
Cho, C. T. and Wenner, H. A. 1973. Monkeypox virus. Bacteriological reviews, 37(1): 1–18. 25 Dennis Jr, J. E. and Schnabel, R. B. 1996. Numerical methods for unconstrained optimization and nonlinear equations, volume 16. Siam
work page 1973
-
[10]
Didier, G., Glatt-Holtz, N. E., Holbrook, A. J., Magee, A. F., and Suchard, M. A. 2024. On the surprising effectiveness of a simple matrix exponential derivative approximation, with application to global sars-cov-2. Proceedings of the National Academy of Sciences , 121(3): e2318989121
work page 2024
-
[11]
Felsenstein, J. 1973. Maximum likelihood and minimum-steps methods for estimating evolutionary trees from data on discrete characters. Syst Biol., 22(3): 240–249
work page 1973
-
[12]
Felsenstein, J. 1981. Evolutionary trees from DNA sequences: a maximum likelihood approach. Journal of Molecular Evolution , 17: 368–376
work page 1981
-
[13]
A., Ji, X., Zhang, Z., Lemey, P., and Suchard, M
Fisher, A. A., Ji, X., Zhang, Z., Lemey, P., and Suchard, M. A. 2021. Relaxed random walks at scale. Systematic Biology , 70(2): 258–267
work page 2021
-
[14]
A., Ji, X., Nishimura, A., Baele, G., Lemey, P., and Suchard, M
Fisher, A. A., Ji, X., Nishimura, A., Baele, G., Lemey, P., and Suchard, M. A. 2023. Shrinkage-based random local clocks with scalable inference. Molecular biology and evolution , 40(11): msad242
work page 2023
-
[15]
Gangavarapu, K., Ji, X., Baele, G., Fourment, M., Lemey, P., Matsen IV, F. A., and Suchard, M. A. 2024. Many-core algorithms for high-dimensional gradients on phylogenetic trees.Bioinformatics, 40(2): btae030
work page 2024
-
[16]
Goldman, N. and Yang, Z. 1994. A codon-based model of nucleotide substitution for protein-coding dna sequences. Molecular biology and evolution , 11(5): 725–736
work page 1994
-
[17]
Guindon, S., Dufayard, J.-F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of phyml 3.0. Systematic biology , 59(3): 307–321
work page 2010
-
[18]
Hasegawa, M., Kishino, H., and Yano, T.-a. 1985. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. Journal of Molecular Evolution , 22(2): 160–174
work page 1985
-
[19]
Hassler, G. W., Magee, A. F., Zhang, Z., Baele, G., Lemey, P., Ji, X., Fourment, M., and Suchard, M. A. 2023. Data integration in bayesian phylogenetics. Annual review of statistics and its application, 10(1): 353–377. 26 H¨ ohna, S., Freyman, W. A., Nolen, Z., Huelsenbeck, J. P., May, M. R., and Moore, B. R. 2019. A bayesian approach for estimating branc...
work page 2023
-
[20]
Holmes, E. C. 2009. The evolution and emergence of RNA viruses . Oxford University Press
work page 2009
-
[21]
P., Larget, B., and Swofford, D
Huelsenbeck, J. P., Larget, B., and Swofford, D. 2000. A compound Poisson process for relaxing the molecular clock. Genetics, 154(4): 1879–1892
work page 2000
-
[22]
Ji, X., Griffing, A., and Thorne, J. L. 2016. A phylogenetic approach finds abundant interlocus gene conversion in yeast. Molecular biology and evolution , 33(9): 2469–2476
work page 2016
-
[23]
Ji, X., Zhang, Z., Holbrook, A., Nishimura, A., Baele, G., Rambaut, A., Lemey, P., and Suchard, M. A. 2020. Gradients do grow on trees: a linear-time o (n)-dimensional gradient for statistical phylogenetics. Molecular biology and evolution , 37(10): 3047–3060
work page 2020
-
[24]
Ji, X., Fisher, A. A., Su, S., Thorne, J. L., Potter, B., Lemey, P., Baele, G., and Suchard, M. A. 2023. Scalable bayesian divergence time estimation with ratio transformations. Systematic Biology , 72(5): 1136–1153
work page 2023
-
[25]
Lemey, P., Rambaut, A., and Pybus, O. G. 2006. Hiv evolutionary dynamics within and among hosts. Aids Rev, 8(3): 125–140
work page 2006
-
[26]
O., Ji, X., Lemey, P., and Suchard, M
Wertheim, J. O., Ji, X., Lemey, P., and Suchard, M. A. 2024. Random-effects substitution models for phylogenetics via scalable gradient approximations. Systematic Biology , 73(3): 562–578
work page 2024
-
[27]
Metropolis, N., Rosenbluth, A. W., Rosenbluth, M. N., Teller, A. H., and Teller, E. 1953. Equation of State Calculations by Fast Computing Machines. The Journal of Chemical Physics , 21(6): 1087–1092
work page 1953
-
[28]
Scheffler, K. 2013. Fubar: a fast, unconstrained bayesian approximation for inferring selection. Molecular biology and evolution , 30(5): 1196–1205. 27
work page 2013
-
[29]
Muse, S. V. and Gaut, B. S. 1994. A likelihood approach for comparing synonymous and nonsyn- onymous nucleotide substitution rates, with application to the chloroplast genome. Molecular biology and evolution , 11(5): 715–724
work page 1994
-
[30]
Najfeld, I. and Havel, T. F. 1995. Derivatives of the matrix exponential and their computation. Advances in applied mathematics , 16(3): 321–375
work page 1995
-
[31]
Neal, R. M. 2011. MCMC using Hamiltonian dynamics. Handbook of Markov Chain Monte Carlo , 2(11). O’Toole, ´A., Neher, R. A., Ndodo, N., Borges, V., Gannon, B., Gomes, J. P., Groves, N., King, D. J., Maloney, D., Lemey, P., et al. 2023. Apobec3 deaminase editing in mpox virus as evidence for sustained human transmission since at least 2016. Science, 382(66...
work page 2011
-
[32]
Kiem, C., and Bedford, T. 2024. Underdetected dispersal and extensive local transmission drove the 2022 mpox epidemic. Cell , 187(6): 1374–1386
work page 2024
-
[33]
Polson, N. G., Scott, J. G., and Windle, J. 2014. The bayesian bridge. Journal of the Royal Statistical Society: Series B (Statistical Methodology) , 76(4): 713–733
work page 2014
-
[34]
Pond, S. L. K. and Frost, S. D. 2005. A genetic algorithm approach to detecting lineage-specific variation in selection pressure. Molecular biology and evolution , 22(3): 478–485
work page 2005
-
[35]
Sherlock, C. 2021. Direct statistical inference for finite markov jump processes via the matrix exponential. Computational Statistics , 36(4): 2863–2887
work page 2021
-
[36]
Stamatakis, A. 2014. Raxml version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics, 30(9): 1312–1313
work page 2014
-
[37]
A., Lemey, P., Baele, G., Ayres, D
Suchard, M. A., Lemey, P., Baele, G., Ayres, D. L., Drummond, A. J., and Rambaut, A. 2018. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol , 4(1): vey016
work page 2018
-
[38]
Tamura, K. and Nei, M. 1993. Estimation of the number of nucleotide substitutions in the control region of mitochondrial dna in humans and chimpanzees. Molecular biology and evolution , 10(3): 512–526. 28
work page 1993
-
[39]
L., Kishino, H., and Painter, I
Thorne, J. L., Kishino, H., and Painter, I. S. 1998. Estimating the rate of evolution of the rate of molecular evolution. Mol Biol Evol. , 15(12): 1647–1657
work page 1998
-
[40]
Wertheim, J. O., Leigh Brown, A. J., Hepler, N. L., Mehta, S. R., Richman, D. D., Smith, D. M., and Kosakovsky Pond, S. L. 2014. The global transmission network of hiv-1. The Journal of infectious diseases, 209(2): 304–313
work page 2014
-
[41]
Yang, Z. 1994. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. Journal of Molecular Evolution , 39(3): 306–314
work page 1994
-
[42]
Yang, Z. 1998. Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Molecular biology and evolution , 15(5): 568–573
work page 1998
-
[43]
Yang, Z. 2007. Paml 4: phylogenetic analysis by maximum likelihood. Molecular biology and evolution, 24(8): 1586–1591
work page 2007
-
[44]
Yang, Z. and Nielsen, R. 1998. Synonymous and nonsynonymous rate variation in nuclear genes of mammals. Journal of molecular evolution , 46: 409–418
work page 1998
-
[45]
Yang, Z. and Nielsen, R. 2002. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Molecular biology and evolution , 19(6): 908–917. 29
work page 2002
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.