Recognition: unknown
Differential Analysis of Microbial Interaction Networks
Pith reviewed 2026-05-08 05:00 UTC · model grok-4.3
The pith
Microbial gene-family networks rewire extensively between male and female patients in three diseases.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
By inferring condition-specific microbial gene-family networks, applying differential network analysis to identify rewired interactions between male and female cohorts, and enriching those interactions for pathways, the framework shows extensive rewiring across inflammatory bowel disease, type 2 diabetes, and atherosclerotic cardiovascular disease. The rewired interactions demonstrate that microbiome alterations involve shifts in community organization in addition to abundance changes, while the enriched pathways uncover latent disease and sex-associated functional mechanisms that are not apparent from the individual networks alone.
What carries the argument
Condition-specific network inference followed by differential network analysis and pathway enrichment on microbial gene-family associations.
If this is right
- Microbiome alterations are shaped by changes in the organization of microbial interactions as well as by changes in abundance.
- Pathway enrichment applied to rewired interactions reveals functional signals that remain invisible when networks from each group are analyzed in isolation.
- The framework can be used to compare microbial functional architecture across other biological conditions or groupings beyond sex.
Where Pith is reading between the lines
- Accounting for sex in microbiome network studies may help explain heterogeneity in disease outcomes between men and women.
- Patterns of rewiring could eventually be tested as additional markers of disease state once the method is applied to larger and more diverse cohorts.
- The same differential approach might be extended to other variables such as age, diet, or treatment response to detect further organizational shifts in microbial communities.
Load-bearing premise
The chosen network inference method recovers true biological interactions and the differential analysis separates genuine condition-specific rewiring from artifacts introduced by the algorithm or data processing.
What would settle it
Repeating the full pipeline with an alternative network inference algorithm yields substantially different sets of rewired interactions, or experimental follow-up shows that the identified rewired edges do not correspond to measurable differences in microbial function or host response between sexes.
Figures

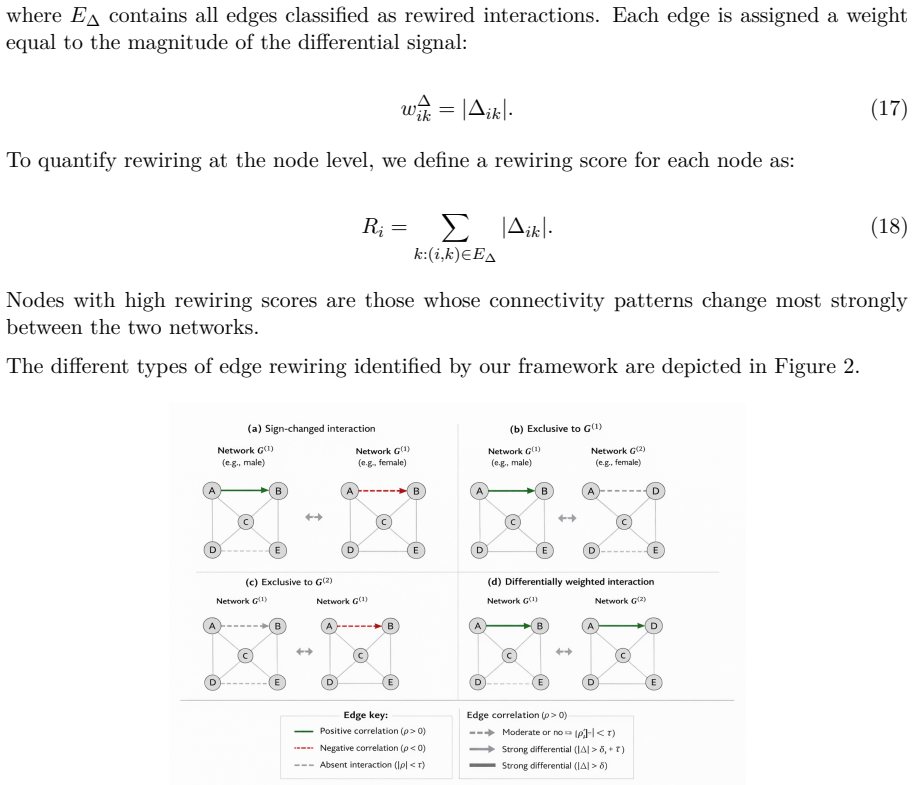
read the original abstract
Microbiome studies increasingly indicate that disease-associated shifts cannot be understood from compositional changes alone. The functional architecture of microbial communities encoded in patterns of association among microbial gene families may reveal how these systems reorganize across biological conditions. Here, we present a network-based framework for characterizing microbiome rewiring across conditions. The approach combines condition-specific network inference, differential network analysis and pathway enrichment to identify interactions that are gained, lost or altered between groups, with a specific focus on sex-dependent differences. We apply the framework to inflammatory bowel disease, type 2 diabetes and atherosclerotic cardiovascular disease, comparing male and female specific microbial gene-family networks within each disease context. Across these settings, differential networks reveal extensive rewiring of microbial functional interactions, suggesting that microbiome alterations are shaped not only by changes in abundance but also by shifts in community organization. Importantly, pathway enrichment of rewired interactions uncovers functional signals that are not apparent from individual networks alone, highlighting latent disease and sex associated mechanisms. Code, data and supplementary information are available on the web site.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces a network-based framework for differential analysis of microbial interaction networks in microbiomes. It combines condition-specific network inference, differential network analysis, and pathway enrichment to identify gained, lost, or altered interactions, with a focus on sex-dependent differences. The framework is applied to inflammatory bowel disease, type 2 diabetes, and atherosclerotic cardiovascular disease, claiming that differential networks reveal extensive rewiring beyond abundance shifts and that pathway enrichment on rewired edges uncovers functional signals not visible in individual networks.
Significance. If the claims hold after rigorous validation, the work could contribute to microbiome research by emphasizing community reorganization over composition alone and by providing a reusable pipeline for sex-stratified differential network analysis. The stated availability of code and data supports reproducibility. However, the absence of any reported validation, statistical controls, or artifact checks in the described framework substantially reduces the current significance, as the headline claims rest on untested premises about inference fidelity.
major comments (3)
- [Abstract] Abstract: The central claim that 'differential networks reveal extensive rewiring of microbial functional interactions' and that 'pathway enrichment of rewired interactions uncovers functional signals that are not apparent from individual networks alone' is presented without any quantitative support, statistical thresholds, error estimates, or baseline comparisons, which are required to establish that the observed patterns exceed what would be expected from the inference procedure itself.
- [Abstract] Abstract: The framework description does not specify the network inference algorithm (e.g., whether it uses Pearson/Spearman correlation, mutual information, or a compositional correction such as SparCC or ANCOM-BC), nor does it indicate how differential edges are identified (e.g., edge-wise permutation tests or stability selection). This omission is load-bearing because standard correlation-based methods on zero-inflated, compositional count data are known to produce spurious condition-dependent edges driven by prevalence and read-depth differences rather than biology.
- [Abstract] Abstract: No mention is made of simulation-based calibration, null-model testing, or sensitivity analyses to confirm that the reported rewiring and enriched pathways are robust to choices in preprocessing, thresholding, or multiple-testing correction. Without these controls, it is impossible to rule out that the 'hidden functional signals' are downstream artifacts of the differential step rather than genuine condition-specific reorganization.
minor comments (1)
- [Abstract] Abstract: The final sentence states that 'Code, data and supplementary information are available on the web site' but provides no URL or repository identifier, which hinders immediate access and reproducibility assessment.
Simulated Author's Rebuttal
We thank the referee for the constructive comments, which identify key areas where the abstract can better convey the methodological details and supporting evidence from the full manuscript. We will revise the abstract and, where appropriate, the main text to incorporate greater specificity on the inference approach, differential testing, quantitative results, and validation procedures.
read point-by-point responses
-
Referee: [Abstract] Abstract: The central claim that 'differential networks reveal extensive rewiring of microbial functional interactions' and that 'pathway enrichment of rewired interactions uncovers functional signals that are not apparent from individual networks alone' is presented without any quantitative support, statistical thresholds, error estimates, or baseline comparisons, which are required to establish that the observed patterns exceed what would be expected from the inference procedure itself.
Authors: We agree that the abstract is high-level and omits the quantitative details provided in the main text and supplements. The manuscript reports the counts of gained, lost, and altered edges (with specific numbers across the three disease cohorts), p-values and FDR thresholds from edge-wise permutation tests, and comparisons against label-shuffled null models showing that observed rewiring exceeds what is expected under the inference procedure. We will revise the abstract to include representative quantitative findings, thresholds, and a brief statement on the null-model comparisons. revision: yes
-
Referee: [Abstract] Abstract: The framework description does not specify the network inference algorithm (e.g., whether it uses Pearson/Spearman correlation, mutual information, or a compositional correction such as SparCC or ANCOM-BC), nor does it indicate how differential edges are identified (e.g., edge-wise permutation tests or stability selection). This omission is load-bearing because standard correlation-based methods on zero-inflated, compositional count data are known to produce spurious condition-dependent edges driven by prevalence and read-depth differences rather than biology.
Authors: The full manuscript specifies SparCC for compositional correction during network inference, followed by stability selection to retain reliable edges and edge-wise permutation testing (with FDR control) to identify differential interactions between conditions. We acknowledge that these details are necessary in the abstract to address potential concerns about artifacts. We will add a concise description of the inference algorithm and differential edge identification method to the revised abstract. revision: yes
-
Referee: [Abstract] Abstract: No mention is made of simulation-based calibration, null-model testing, or sensitivity analyses to confirm that the reported rewiring and enriched pathways are robust to choices in preprocessing, thresholding, or multiple-testing correction. Without these controls, it is impossible to rule out that the 'hidden functional signals' are downstream artifacts of the differential step rather than genuine condition-specific reorganization.
Authors: The manuscript includes sensitivity analyses (varying preprocessing, thresholding, and multiple-testing corrections) and null-model testing via label permutation, reported in the supplementary materials, to demonstrate robustness of the rewiring and pathway enrichment results. We agree that explicit reference to these controls should appear in the abstract. We will revise the abstract to note the validation approaches and will expand the main text with a brief summary of the sensitivity checks. revision: partial
Circularity Check
No circularity in conceptual framework
full rationale
The manuscript presents a high-level methodological framework combining condition-specific network inference, differential analysis, and pathway enrichment, then applies it empirically to microbiome data from three diseases. No equations, parameter fits, derivation chains, or mathematical predictions are described that could reduce to inputs by construction. Claims rest on observed rewiring patterns and enrichment results rather than tautological re-statements of fitted quantities or self-citation load-bearing premises. The approach is self-contained as a conceptual pipeline with external data application and does not invoke uniqueness theorems, ansatzes, or renamed known results.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
T. C. Fung, C. A. Olson, E. Y. Hsiao, Interactions between the microbiota, immune and nervous systems in health and disease, Nature neuroscience 20 (2) (2017) 145–155
2017
-
[2]
L. Lin, J. Zhang, Role of intestinal microbiota and metabolites on gut homeostasis and human diseases, BMC immunology 18 (1) (2017) 2
2017
-
[3]
Manor, C
O. Manor, C. L. Dai, S. A. Kornilov, B. Smith, N. D. Price, J. C. Lovejoy, S. M. Gibbons, A. T. Magis, Health and disease markers correlate with gut microbiome composition across thousands of people, Nature communications 11 (1) (2020) 5206
2020
-
[4]
J. M. Pickard, M. Y. Zeng, R. Caruso, G. Núñez, Gut microbiota: Role in pathogen colonization, immune responses, and inflammatory disease, Immunological reviews 279 (1) (2017) 70–89
2017
-
[5]
Gutiérrez-Sacristán, A
A. Gutiérrez-Sacristán, A. Serret-Larmande, M. R. Hutch, C. Sáez, B. J. Aronow, S. Bhat- nagar, C.-L. Bonzel, T. Cai, B. Devkota, D. A. Hanauer, et al., Hospitalizations associated with mental health conditions among adolescents in the us and france during the covid-19 pandemic, JAMA network open 5 (12) (2022) e2246548
2022
-
[6]
Agapito, M
G. Agapito, M. Cannataro, P. H. Guzzi, F. Marozzo, D. Talia, P. Trunfio, Cloud4snp: dis- tributed analysis of snp microarray data on the cloud, in: Proceedings of the International Conference on Bioinformatics, Computational Biology and Biomedical Informatics, 2013, pp. 468–475
2013
-
[7]
Srinivasan, A
S. Srinivasan, A. Jnana, T. S. Murali, Modeling microbial community networks: methods and tools for studying microbial interactions, Microbial ecology 87 (1) (2024) 56
2024
-
[8]
J. Kim, I. Kim, S. K. Han, J. U. Bowie, S. Kim, Network rewiring is an important mecha- nism of gene essentiality change, Scientific reports 2 (1) (2012) 900
2012
-
[9]
P. H. Guzzi, A. Roy, M. Milano, P. Veltri, Non parametric differential network analysis: a tool for unveiling specific molecular signatures, BMC bioinformatics 25 (1) (2024) 359. 12
2024
-
[10]
Faust, J
K. Faust, J. F. Sathirapongsasuti, J. Izard, N. Segata, D. Gevers, J. Raes, C. Huttenhower, Microbial co-occurrence relationships in the human microbiome, PLoS computational bi- ology 8 (7) (2012) e1002606
2012
-
[11]
Weiss, W
S. Weiss, W. Van Treuren, C. Lozupone, K. Faust, J. Friedman, Y. Deng, L. C. Xia, Z. Z. Xu, L. Ursell, E. J. Alm, et al., Correlation detection strategies in microbial data sets vary widely in sensitivity and precision, The ISME journal 10 (7) (2016) 1669–1681
2016
-
[12]
J. M. Macklaim, A. D. Fernandes, J. M. Di Bella, J.-A. Hammond, G. Reid, G. B. Gloor, Comparative meta-rna-seq of the vaginal microbiota and differential expression by lacto- bacillus iners in health and dysbiosis, Microbiome 1 (1) (2013) 12
2013
-
[13]
Hou, Z.-X
K. Hou, Z.-X. Wu, X.-Y. Chen, J.-Q. Wang, D. Zhang, C. Xiao, D. Zhu, J. B. Koya, L. Wei, J. Li, et al., Microbiota in health and diseases, Signal transduction and targeted therapy 7 (1) (2022) 135
2022
-
[14]
G. B. Gloor, J. M. Macklaim, V. Pawlowsky-Glahn, J. J. Egozcue, Microbiome datasets are compositional: and this is not optional, Frontiers in microbiology 8 (2017) 2224
2017
-
[15]
Z.D.Kurtz, C.L.Müller, E.R.Miraldi, D.R.Littman, M.J.Blaser, R.A.Bonneau, Sparse and compositionally robust inference of microbial ecological networks, PLoS computational biology 11 (5) (2015) e1004226
2015
-
[16]
J. G. Markle, D. N. Frank, S. Mortin-Toth, C. E. Robertson, L. M. Feazel, U. Rolle- Kampczyk, M. Von Bergen, K. D. McCoy, A. J. Macpherson, J. S. Danska, Sex differ- ences in the gut microbiome drive hormone-dependent regulation of autoimmunity, Science 339 (6123) (2013) 1084–1088
2013
-
[17]
Ahmed, J
S. Ahmed, J. D. Spence, Sex differences in the intestinal microbiome: interactions with risk factors for atherosclerosis and cardiovascular disease, Biology of sex Differences 12 (1) (2021) 35
2021
-
[18]
Jašarević, K
E. Jašarević, K. E. Morrison, T. L. Bale, Sex differences in the gut microbiome–brain axis across the lifespan, Philosophical Transactions of the Royal Society B: Biological Sciences 371 (1688) (2016)
2016
-
[19]
Guven-Maiorov, C.-J
E. Guven-Maiorov, C.-J. Tsai, R. Nussinov, Structural host-microbiota interaction net- works, PLoS computational biology 13 (10) (2017) e1005579
2017
-
[20]
Friedman, E
J. Friedman, E. J. Alm, Inferring correlation networks from genomic survey data (2012)
2012
-
[21]
Peschel, C
S. Peschel, C. L. Müller, E. Von Mutius, A.-L. Boulesteix, M. Depner, Netcomi: network construction and comparison for microbiome data in r, Briefings in bioinformatics 22 (4) (2021) bbaa290
2021
-
[22]
J. L. Espinoza, M. Torralba, P. Leong, R. Saffery, M. Bockmann, C. Kuelbs, S. Singh, T.Hughes, J.M.Craig, K.E.Nelson, etal., Differentialnetworkanalysisoforalmicrobiome metatranscriptomes identifies community scale metabolic restructuring in dental caries, PNAS nexus 1 (5) (2022) pgac239
2022
-
[23]
S. Ahn, S. Datta, Differential network connectivity analysis for microbiome data adjusted for clinical covariates using jackknife pseudo-values, BMC bioinformatics 25 (1) (2024) 117
2024
-
[24]
McGregor, A
K. McGregor, A. Labbe, C. M. Greenwood, Mdine: a model to estimate differential co- occurrence networks in microbiome studies, Bioinformatics 36 (6) (2020) 1840–1847
2020
- [25]
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.