Bayesian genome-wide clustering and variable selection of transcriptomic data via rank-based mixtures
Pith reviewed 2026-06-27 23:55 UTC · model grok-4.3
The pith
A new rank-based Bayesian mixture model performs joint clustering and variable selection on ultra-high-dimensional transcriptomic data.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The paper establishes that the lower-dimensional Bayesian Mallows Model Mixture (lowBM3) provides the first rank-based extension of the Bayesian Mallows model capable of jointly performing clustering and variable selection on ultra-high-dimensional ranking data. This framework simultaneously accounts for sample heterogeneity, performs unsupervised parameter estimation, and conducts model selection while remaining computationally feasible for transcriptomic applications. A companion postprocessing procedure yields summaries of the discrete posterior distributions for the consensus ranking and the variable selector. Simulation studies confirm the method's performance, and an application to bul
What carries the argument
The lower-dimensional Bayesian Mallows Model Mixture (lowBM3), a rank-based mixture that extends the Bayesian Mallows model to lower dimensions for simultaneous clustering of samples and selection of variables from high-dimensional rankings.
If this is right
- The approach scales Bayesian rank-based inference to ultra-high-dimensional settings previously inaccessible to standard BMM.
- It supplies full posterior uncertainty quantification for both cluster assignments and the selected variables.
- Unsupervised clustering and variable selection occur together without requiring separate preprocessing steps.
- Postprocessing yields interpretable summaries of the discrete posterior distributions over rankings and selectors.
- The framework supports signature discovery in cancer genomics by clustering patients and identifying relevant genes from ranked expression data.
Where Pith is reading between the lines
- If lowBM3 succeeds on transcriptomic rankings, the same lower-dimensional reduction could be tested on other high-dimensional ranking problems such as preference data or sports rankings.
- The method's robustness to non-normality might allow direct comparison against traditional mixture models on the same datasets to quantify gains from the rank-based formulation.
- Successful genome-wide application suggests the model could be adapted for integrative analysis across multiple omics layers if rankings can be aligned.
- Scalability gains open the possibility of applying the method to longitudinal or multi-condition transcriptomic experiments where dimensions are even larger.
Load-bearing premise
The Mallows model structure stays appropriate when extended to joint clustering and variable selection in ultra-high-dimensional transcriptomic ranking data without creating prohibitive computational or modeling biases.
What would settle it
Simulation results or the breast cancer application in which lowBM3 fails to recover known structure, produces unstable variable selections, or becomes computationally infeasible on typical genome-wide datasets would show the central claim does not hold.
Figures
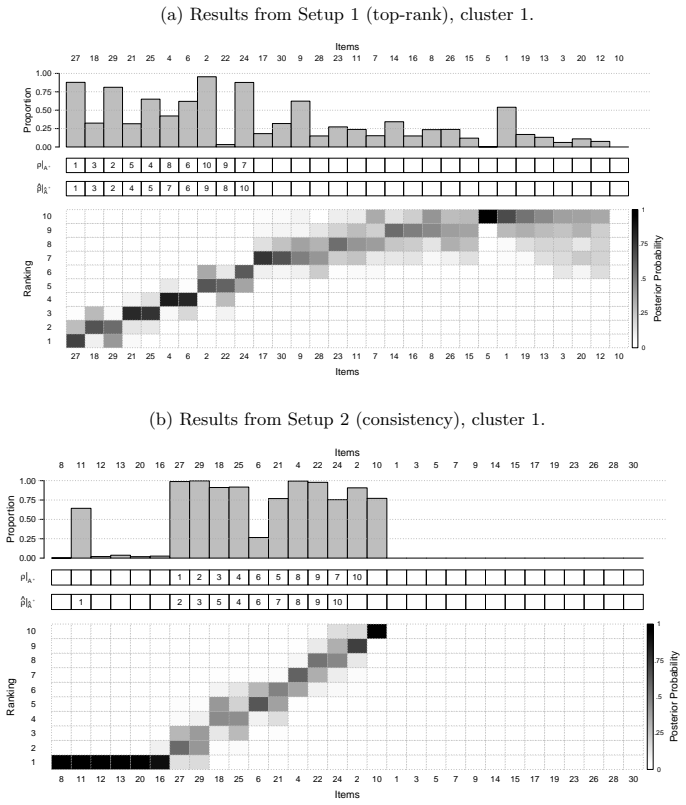
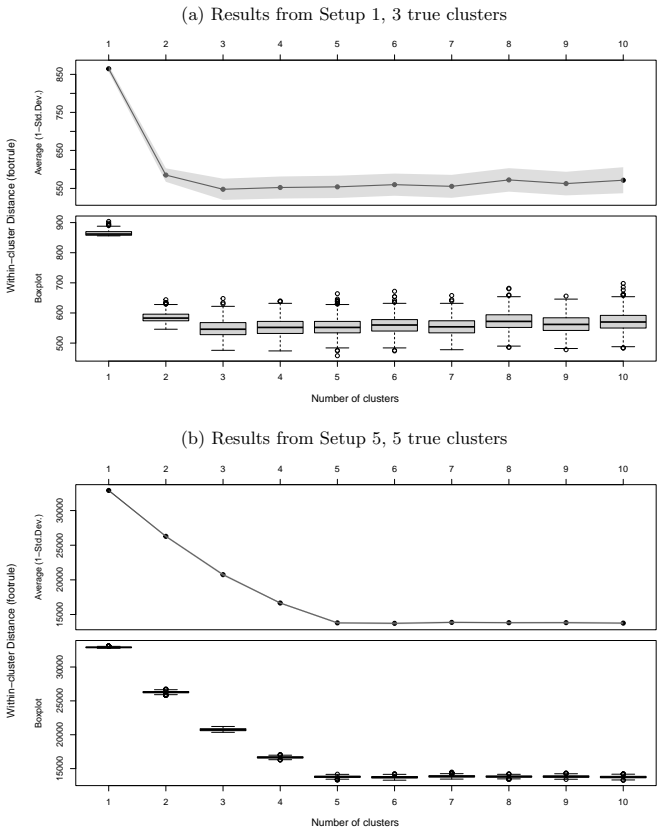
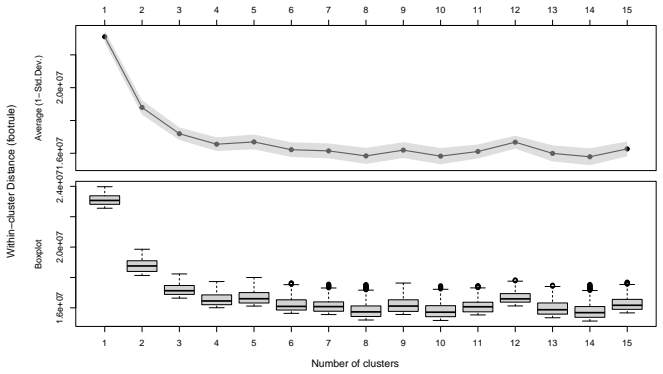
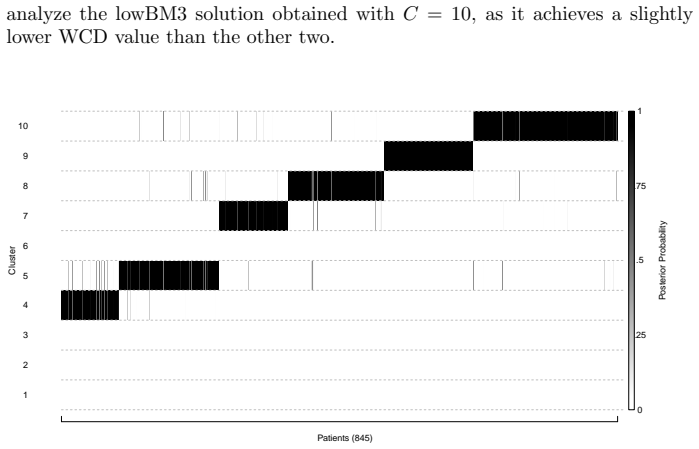
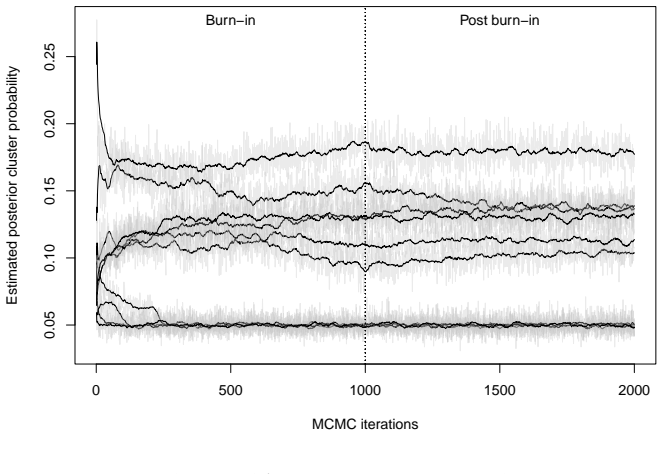
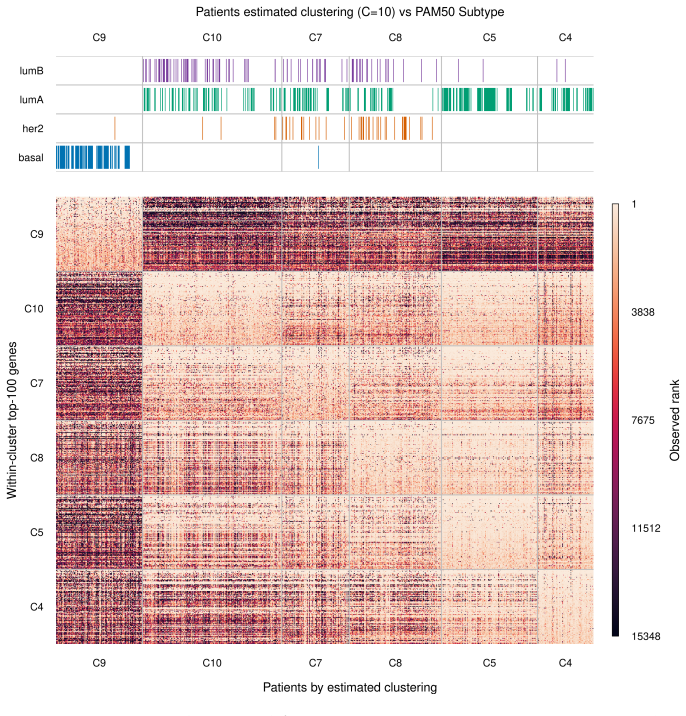
read the original abstract
With the increasing availability of ranking data, there has been a growing demand for appropriate unsupervised rank-based inferential frameworks capable of handling high-dimensional datasets and providing uncertainty quantification for all estimates. Rank-based methods have also seen a growing popularity in -omics pipelines, as ranking continuous measurements provides a robust means of handling non-normally distributed data. The Bayesian Mallows model (BMM) has emerged as a promising choice because of its adaptability to various types of ranking data and its flexible framework, integrating cluster-wise rank aggregation with inference at the individual level. However, the scalability of BMM to ultra-high-dimensional settings, such as -omics analyses, has remained limited. The present paper addresses this issue by introducing the first rank-based model generalizing BMM to jointly handle clustering and variable selection, namely the lower-dimensional Bayesian Mallows Model Mixture (lowBM3). The proposed method provides a novel Bayesian framework that simultaneously handles heterogeneity in the sample, unsupervised parameter estimation, and model selection in a scalable manner for ultra-high-dimensional data. Additionally, a companion postprocessing framework is introduced to provide posterior summaries of the discrete posterior distributions of both the consensus ranking and the variable selector. Simulation studies are performed to assess the performance of the method. The usefulness of the method is also shown in an application to signature discovery for cancer genomics, where RNA-seq bulk gene expression data obtained from breast cancer patients are clustered genome-wide.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces the lower-dimensional Bayesian Mallows Model Mixture (lowBM3) as the first rank-based generalization of the Bayesian Mallows Model (BMM) that jointly performs clustering and variable selection for ultra-high-dimensional transcriptomic ranking data. It claims to provide a scalable Bayesian framework for handling sample heterogeneity, unsupervised estimation, and model selection, along with a postprocessing step for posterior summaries of consensus rankings and variable selectors; performance is assessed via simulation studies and demonstrated on breast cancer RNA-seq data for signature discovery.
Significance. If the scalability and joint inference claims hold with appropriate error control, the work would offer a useful extension of rank-based mixture models to genome-wide omics settings where continuous data are converted to rankings for robustness, supplying full posterior uncertainty quantification that is currently limited in high-dimensional BMM applications.
major comments (2)
- [§3] §3 (Model specification): the lowBM3 likelihood and prior structure for the joint clustering-variable selection indicator must be shown explicitly; without the precise form of the dimension-reduction step and how the Mallows distance is computed only on the selected variables, it is unclear whether the claimed scalability follows from the model or from an unstated approximation.
- [§5] §5 (Simulation studies): the reported recovery rates and clustering metrics for the variable selector and consensus ranking are not accompanied by any quantification of Monte Carlo error or sensitivity to the choice of the concentration parameter; this weakens the claim that the method reliably outperforms existing BMM implementations in ultra-high dimensions.
minor comments (2)
- [Abstract] The abstract states that simulations assess performance but does not report any numerical values or baselines; adding a short table of key metrics would improve readability.
- [§2] Notation for the variable selector indicator and the reduced ranking vector should be introduced once in §2 and used consistently thereafter to avoid ambiguity in the postprocessing description.
Simulated Author's Rebuttal
We thank the referee for their constructive comments. We address each major comment below and have revised the manuscript accordingly to improve clarity and reporting.
read point-by-point responses
-
Referee: [§3] §3 (Model specification): the lowBM3 likelihood and prior structure for the joint clustering-variable selection indicator must be shown explicitly; without the precise form of the dimension-reduction step and how the Mallows distance is computed only on the selected variables, it is unclear whether the claimed scalability follows from the model or from an unstated approximation.
Authors: We thank the referee for this suggestion. While the model was introduced in Section 3, we agree that greater explicitness is warranted. In the revised manuscript we have added the full likelihood expression (new Equation 2) together with the prior on the joint clustering and variable-selection indicator vector. The dimension-reduction step is now stated precisely: the Mallows distance is computed exclusively on the coordinates where the selection indicator equals one, with no further approximation. This explicit construction is what yields the reported scalability; the revised text includes a short paragraph clarifying the computational consequence of the restricted distance. revision: yes
-
Referee: [§5] §5 (Simulation studies): the reported recovery rates and clustering metrics for the variable selector and consensus ranking are not accompanied by any quantification of Monte Carlo error or sensitivity to the choice of the concentration parameter; this weakens the claim that the method reliably outperforms existing BMM implementations in ultra-high dimensions.
Authors: We accept this criticism. The revised Section 5 now reports Monte Carlo standard errors for every recovery rate and clustering metric, computed from ten independent MCMC runs with different random seeds. We have also added a sensitivity study that varies the concentration parameter over a grid and tabulates the resulting changes in performance; the outcomes remain consistent with the original claims. These additions directly address the concern about error control and robustness. revision: yes
Circularity Check
No significant circularity detected
full rationale
The paper introduces lowBM3 as a novel generalization of the Bayesian Mallows Model (BMM) to jointly perform clustering and variable selection on ultra-high-dimensional transcriptomic ranking data. The abstract describes this as addressing prior scalability limits of BMM through a new Bayesian framework, with simulation studies and a cancer genomics application. No derivation steps, equations, or claims are presented that reduce by construction to fitted parameters, self-citations, or renamed inputs; the central contribution is framed as an independent modeling extension rather than a re-expression of prior results. The work is self-contained against external benchmarks, with the Mallows structure treated as a modeling choice supported by prior literature.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption Mallows model is suitable base for rank data in transcriptomics
- ad hoc to paper Joint clustering and variable selection can be performed scalably in Bayesian framework for ultra-high dimensions
invented entities (1)
-
lowBM3 model
no independent evidence
Reference graph
Works this paper leans on
-
[1]
Rank Discrim- inants for Predicting Phenotypes from RNA Expression.The Annals of Applied Statistics, 8(3):1469–1491, 2014
Bahman Afsari, Ulisses Braga-Neto, and Donald Geman. Rank Discrim- inants for Predicting Phenotypes from RNA Expression.The Annals of Applied Statistics, 8(3):1469–1491, 2014
2014
-
[2]
Deciphering signatures of mutational processes operative in human cancer.Cell reports, 3(1):246–259, 2013
Ludmil Alexandrov, Serena Nik-Zainal, David Wedge, Peter Campbell, and Michael Stratton. Deciphering signatures of mutational processes operative in human cancer.Cell reports, 3(1):246–259, 2013
2013
-
[3]
A tutorial on adaptive mcmc
Christophe Andrieu and Johannes Thoms. A tutorial on adaptive mcmc. Statistics and computing, 18(4):343–373, 2008
2008
-
[4]
MOFA+: a statistical framework for comprehensive integration of multi-modal single-cell data
Ricard Argelaguet, Damien Arnol, Danila Bredikhin, Yonatan Deloro, Britta Velten, John C Marioni, and Oliver Stegle. MOFA+: a statistical framework for comprehensive integration of multi-modal single-cell data. Genome Biology, 21(1):111, 2020
2020
-
[5]
Integrative clustering reveals a novel split in the lumi- nal A subtype of breast cancer with impact on outcome.Breast Cancer Research, 19(1):44, 2017
Miriam Aure, Valeria Vitelli, Sandra Jernstr¨ om, Surendra Kumar, Marit Krohn, Eldri Due, Tonje Haukaas, Suvi-Katri Leivonen, Hans Vollan, Tor- ben L¨ uders, Einar Rødland, Charles Vaske, Wei Zhao, Elen Møller, Silje Nord, Guro Giskeødeg˚ ard, Tone Bathen, Carlos Caldas, Trine Tramm, and Kristine Sahlberg. Integrative clustering reveals a novel split in t...
2017
-
[6]
Badgeley, Stuart C
Marcus A. Badgeley, Stuart C. Sealfon, and Maria D. Chikina. Hybrid Bayesian-rank integration approach improves the predictive power of ge- nomic dataset aggregation.Bioinformatics, 31(2):209–215, 2015
2015
-
[7]
Understanding uncertainty in bayesian cluster analysis.arXiv preprint arXiv:2506.16295, 2025
Cecilia Balocchi and Sara Wade. Understanding uncertainty in bayesian cluster analysis.arXiv preprint arXiv:2506.16295, 2025
-
[8]
PAM50 breast cancer subtyping 34 by RT-qPCR and concordance with standard clinical molecular markers
Roy RL Bastien, ´Alvaro Rodr´ ıguez-Lescure, Mark TW Ebbert, Aleix Prat, Blanca Mun´ arriz, Leslie Rowe, Patricia Miller, Manuel Ruiz-Borrego, Daniel Anderson, Bradley Lyons, et al. PAM50 breast cancer subtyping 34 by RT-qPCR and concordance with standard clinical molecular markers. BMC Medical Genomics, 5(1):44, 2012
2012
-
[9]
Adaptive stereographic mcmc.arXiv preprint arXiv:2408.11780, 2024
Cameron Bell, Krzystof Latuszy´ nski, and Gareth O Roberts. Adaptive stereographic mcmc.arXiv preprint arXiv:2408.11780, 2024
-
[10]
Chen and Daniela M
Yiqun T. Chen and Daniela M. Witten. Selective inference for k-means clustering.Journal of Machine Learning Research, 24(152):1–41, 2023
2023
-
[11]
Stability post-processing for items im- portance in preference learning via the bayesian mallows model
Luca Coraggio and Valeria Vitelli. Stability post-processing for items im- portance in preference learning via the bayesian mallows model. In An- tonio D’Ambrosio, Mark de Rooij, Kim De Roover, Carmela Iorio, and Michele La Rocca, editors,Supervised and Unsupervised Statistical Data Analysis, Studies in Classification, Data Analysis, and Knowledge Organi-...
2025
-
[12]
The genomic and transcriptomic ar- chitecture of 2,000 breast tumours reveals novel subgroups.Nature, 486(7403):346–352, 2012
Christina Curtis, Sohrab P Shah, Suet-Feung Chin, Gulisa Turashvili, Os- car M Rueda, Mark J Dunning, Doug Speed, Andy G Lynch, Shamith Samarajiwa, Yinyin Yuan, et al. The genomic and transcriptomic ar- chitecture of 2,000 breast tumours reveals novel subgroups.Nature, 486(7403):346–352, 2012
2012
-
[13]
Bayesian aggregation of order-based rank data.Journal of the American Statistical Association, 109(507):1023–1039, 2014
Ke Deng, Simeng Han, Kate Li, and Jun Liu. Bayesian aggregation of order-based rank data.Journal of the American Statistical Association, 109(507):1023–1039, 2014
2014
-
[14]
Vol- ume 11 of Lecture Notes - Monograph Series
Persi Diaconis.Group Representations in Probability and Statistics. Vol- ume 11 of Lecture Notes - Monograph Series. Institute of Mathematical Statistics, Hayward, CA, USA, 1988
1988
-
[15]
Rank-based bayesian variable selection for genome-wide transcriptomic analyses.Statis- tics in Medicine, 41(23):4532–4553, 2022
Emilie Eliseussen, Thomas Fleischer, and Valeria Vitelli. Rank-based bayesian variable selection for genome-wide transcriptomic analyses.Statis- tics in Medicine, 41(23):4532–4553, 2022
2022
-
[16]
Systematic bias in genomic classifi- cation due to contaminating non-neoplastic tissue in breast tumor samples
Fathi Elloumi, Zhiyuan Hu, Yan Li, Joel S Parker, Margaret L Gulley, Keith D Amos, and Melissa A Troester. Systematic bias in genomic classifi- cation due to contaminating non-neoplastic tissue in breast tumor samples. BMC Medical Genomics, 4(1):54, 2011
2011
-
[17]
Dna methylation at enhancers identifies distinct breast cancer lineages.Nature Communications, 8(1):1379, 2017
Thomas Fleischer, Xavier Tekpli, Anthony Mathelier, Shixiong Wang, Daniel Nebdal, Hari P Dhakal, Kristine Kleivi Sahlberg, Ellen Schlichting, Anne-Lise Børresen-Dale, et al. Dna methylation at enhancers identifies distinct breast cancer lineages.Nature Communications, 8(1):1379, 2017
2017
-
[18]
M. A. Fligner and J. S. Verducci. Distance Based Ranking Models.Journal of the Royal Statistical Society: Series B (Methodological), 48(3):359–369, 1986
1986
-
[19]
Variable selection methods for model-based clustering.Statistics Surveys, 12:18–65, 2018
Michael Fop and Thomas Brendan Murphy. Variable selection methods for model-based clustering.Statistics Surveys, 12:18–65, 2018. 35
2018
-
[20]
Selective inference for hierarchical clustering.Journal of the American Statistical Association, 119(545):332–342, 2024
Lucy L Gao, Jacob Bien, and Daniela Witten. Selective inference for hierarchical clustering.Journal of the American Statistical Association, 119(545):332–342, 2024
2024
-
[21]
Uncovering clinically relevant breast cancer subtypes biomarkers using integrative bioinformatics and machine learning approaches.Biomarkers, pages 1–12, 2026
Prashansha Goel and Nilofer Shaikh. Uncovering clinically relevant breast cancer subtypes biomarkers using integrative bioinformatics and machine learning approaches.Biomarkers, pages 1–12, 2026. Accepted author ver- sion posted online: 09 Mar 2026
2026
-
[22]
Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin
Katherine A Hoadley, Christina Yau, Denise M Wolf, Andrew D Cherniack, David Tamborero, Sam Ng, Max DM Leiserson, Beifang Niu, Michael D McLellan, Vladislav Uzunangelov, et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell, 158(4):929–944, 2014
2014
-
[23]
RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome.BMC Bioinformatics, 12(1):323, 2011
Bo Li and Colin N Dewey. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome.BMC Bioinformatics, 12(1):323, 2011
2011
-
[24]
A sparse negative binomial mixture model for clustering RNA-seq count data.Biostatistics, 24(1):68–84, 2023
Yujia Li, Tanbin Rahman, Tianzhou Ma, Lu Tang, and George C Tseng. A sparse negative binomial mixture model for clustering RNA-seq count data.Biostatistics, 24(1):68–84, 2023
2023
-
[25]
Model-based learning from preference data.Annual Review of Statistics and Its Application, 6(1):329–354, 2019
Qinghua Liu, Marta Crispino, Ida Scheel, Valeria Vitelli, and Arnoldo Frigessi. Model-based learning from preference data.Annual Review of Statistics and Its Application, 6(1):329–354, 2019
2019
-
[26]
Effective Sampling and Learning for Mal- lows Models with Pairwise-Preference Data.Journal of Machine Learning Research, 15(117):3963–4009, 2014
Tyler Lu and Craig Boutilier. Effective Sampling and Learning for Mal- lows Models with Pairwise-Preference Data.Journal of Machine Learning Research, 15(117):3963–4009, 2014
2014
-
[27]
Luce.Individual choice behavior: A theoretical analysis
Robert D. Luce.Individual choice behavior: A theoretical analysis. Wiley, New York, NY, USA, 1959
1959
-
[28]
Non-null ranking models
Colin L Mallows. Non-null ranking models. i.Biometrika, 44(1/2):114–130, 1957
1957
-
[29]
An Exponential Model for Infinite Rankings
Marina Meil˘ a and Le Bao. An Exponential Model for Infinite Rankings. Journal of Machine Learning Research, 11(113):3481–3518, 2010
2010
-
[30]
Dirichlet Process Mixtures of Generalized Mallows Models
Marina Meil˘ a and Harr Chen. Dirichlet Process Mixtures of Generalized Mallows Models. InProceedings of the Twenty-Sixth Conference on Uncer- tainty in Artificial Intelligence, UAI’10, pages 358–367, Arlington, Virginia, USA, 2010. AUAI Press
2010
-
[31]
A fully bayesian latent variable model for integrative clustering analysis of multi-type omics data.Biostatistics, 19(1):71–86, 05 2017
Qianxing Mo, Ronglai Shen, Cui Guo, Marina Vannucci, Keith S Chan, and Susan G Hilsenbeck. A fully bayesian latent variable model for integrative clustering analysis of multi-type omics data.Biostatistics, 19(1):71–86, 05 2017. 36
2017
-
[32]
PGC-1 alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human di- abetes.Nature Genetics, 34:267–73, 2003
Vamsi Mootha, Cecilia Lindgren, Karl-Fredrik Eriksson, Aravind Subrama- nian, Smita Sihag, Joseph Lehar, Pere Puigserver, Emma Nilsson, Martin Ridderstr˚ ale, Esa Laurila, Nicholas Houstis, Mark Daly, Nick Patterson, Jill Mesirov, Todd Golub, Pablo Tamayo, Bruce Spiegelman, Eric Lander, Joel Hirschhorn, and Leif Groop. PGC-1 alpha-responsive genes involve...
2003
-
[33]
Spike-and-slab lasso biclustering.The Annals of Applied Statistics, 15(1):148–173, 2021
Gemma E Moran, Veronika Roˇ ckov´ a, and Edward I George. Spike-and-slab lasso biclustering.The Annals of Applied Statistics, 15(1):148–173, 2021
2021
-
[34]
Comparison of sparse biclustering algorithms for gene expression datasets.Briefings in bioinformatics, 22(6):bbab140, 2021
Kath Nicholls and Chris Wallace. Comparison of sparse biclustering algorithms for gene expression datasets.Briefings in bioinformatics, 22(6):bbab140, 2021
2021
-
[35]
Parker, Michael Mullins, Maggie Chon U Cheang, Samuel C Y Le- ung, David Voduc, Tammi L
Joel S. Parker, Michael Mullins, Maggie Chon U Cheang, Samuel C Y Le- ung, David Voduc, Tammi L. Vickery, Sherri R. Davies, Christiane M.-R. Fauron, Xiaping He, Zhiyuan Hu, John F. Quackenbush, Inge J. Stijle- man, Juan P. Palazzo, J. S. Marron, Andrew B. Nobel, Elaine R. Mardis, Torsten O. Nielsen, Matthew J. Ellis, Charles M. Perou, and Philip S. Bernar...
2009
-
[36]
Molecular portraits of human breast tumours.Nature, 406(6797):747–752, 2000
Charles M Perou, Therese Sørlie, Michael B Eisen, Matt Van De Rijn, Stefanie S Jeffrey, Christian A Rees, Jonathan R Pollack, Douglas T Ross, Hilde Johnsen, Lars A Akslen, et al. Molecular portraits of human breast tumours.Nature, 406(6797):747–752, 2000
2000
-
[37]
Plackett
Robin L. Plackett. The Analysis of Permutations.Journal of the Royal Statistical Society: Series C (Applied Statistics), 24(2):193–202, 1975
1975
-
[38]
Clinical implications of the intrinsic molecular subtypes of breast cancer.The Breast, 24:S26–S35, 2015
Aleix Prat, Estela Pineda, Barbara Adamo, Patricia Galv´ an, Aranzazu Fern´ andez, Lydia Gaba, Marc D´ ıez, Margarita Viladot, Ana Arance, and Montserrat Mu˜ noz. Clinical implications of the intrinsic molecular subtypes of breast cancer.The Breast, 24:S26–S35, 2015
2015
-
[39]
Network-based prioritization of cancer genes by integrative ranks from multi-omics data.Computers in Biology and Medicine, 119:103692, 2020
Haixia Shang and Zhi-Ping Liu. Network-based prioritization of cancer genes by integrative ranks from multi-omics data.Computers in Biology and Medicine, 119:103692, 2020
2020
-
[40]
BayesMallows: An R Package for the Bayesian Mallows Model.The R Journal, 12(1):324–342, 2020
Øystein Sørensen, Marta Crispino, Qinghua Liu, and Valeria Vitelli. BayesMallows: An R Package for the Bayesian Mallows Model.The R Journal, 12(1):324–342, 2020
2020
-
[41]
Mootha, Sayan Mukher- jee, Benjamin L
Aravind Subramanian, Pablo Tamayo, Vamsi K. Mootha, Sayan Mukher- jee, Benjamin L. Ebert, Michael A. Gillette, Amanda Paulovich, Scott L. Pomeroy, Todd R. Golub, Eric S. Lander, and Jill P. Mesirov. Gene set enrichment analysis: A knowledge-based approach for interpreting genome- wide expression profiles.Proceedings of the National Academy of Sciences, 10...
2005
-
[42]
Perou, Robert Tibshirani, Turid Aas, Stephanie Geisler, Hilde Johnsen, Trevor Hastie, Michael B
Therese Sørlie, Charles M. Perou, Robert Tibshirani, Turid Aas, Stephanie Geisler, Hilde Johnsen, Trevor Hastie, Michael B. Eisen, Matt van de Rijn, Stefanie S. Jeffrey, Thor Thorsen, Hanne Quist, John C. Matese, Patrick O. Brown, David Botstein, Per Eystein Lønning, and Anne-Lise Børresen- Dale. Gene expression patterns of breast carcinomas distinguish t...
2001
-
[43]
Bayesian variable selection in clustering high-dimensional data.Journal of the American Statistical Association, 100(470):602–617, 2005
Mahlet G Tadesse, Naijun Sha, and Marina Vannucci. Bayesian variable selection in clustering high-dimensional data.Journal of the American Statistical Association, 100(470):602–617, 2005
2005
-
[44]
Olga Troyanskaya, Michael Cantor, Gavin Sherlock, Pat Brown, Trevor Hastie, Robert Tibshirani, David Botstein, and Russ B. Altman. Missing value estimation methods for DNA microarrays.Bioinformatics, 17(6):520– 525, 2001
2001
-
[45]
Probabilistic preference learning with the mallows rank model
Valeria Vitelli, Øystein Sørensen, Marta Crispino, Arnoldo Frigessi, and Elja Arjas. Probabilistic preference learning with the mallows rank model. Journal of Machine Learning Research, 18(158):1–49, 2018
2018
-
[46]
Bayesian Cluster Analysis: Point Esti- mation and Credible Balls (with Discussion).Bayesian Analysis, 13(2):559– 626, 2018
Sara Wade and Zoubin Ghahramani. Bayesian Cluster Analysis: Point Esti- mation and Credible Balls (with Discussion).Bayesian Analysis, 13(2):559– 626, 2018
2018
-
[47]
MapSplice: Accurate mapping of RNA-seq reads for splice junction discovery.Nucleic Acids Research, 38(18):e178, 2010
Kai Wang, Darshan Singh, Zheng Zeng, Stephen Coleman, Yan Huang, Gleb Savich, Xiaping He, Piotr Mieczkowski, Sara Grimm, Charles Perou, James MacLeod, Derek Chiang, Jan Prins, and Jinze Liu. MapSplice: Accurate mapping of RNA-seq reads for splice junction discovery.Nucleic Acids Research, 38(18):e178, 2010
2010
-
[48]
Breast cancer molecular profiling with single sample predictors: a retrospective analysis
Britta Weigelt, Alan Mackay, Roger A’hern, Rachael Natrajan, David SP Tan, Mitch Dowsett, Alan Ashworth, and Jorge S Reis-Filho. Breast cancer molecular profiling with single sample predictors: a retrospective analysis. The Lancet Oncology, 11(4):339–349, 2010
2010
-
[49]
Witten and Robert Tibshirani
Daniela M. Witten and Robert Tibshirani. A framework for feature se- lection in clustering.Journal of the American Statistical Association, 105(490):713–726, 2010. PMID: 20811510
2010
-
[50]
Michael H. Zhu. Sample adaptive mcmc.Advances in Neural Information Processing Systems, 32, 2019. 38 Supplementary Material Bayesian genome-wide clustering and variable selection of transcriptomic data via rank-based mixtures A Noisy data generating processes We devise two additional data generating processes (DGPs) that add noise to the consistency DGP. ...
2019
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.