A benchmark suite of intracellular Boolean model variants and multiscale simulations for computational biology
Pith reviewed 2026-06-26 21:42 UTC · model grok-4.3
The pith
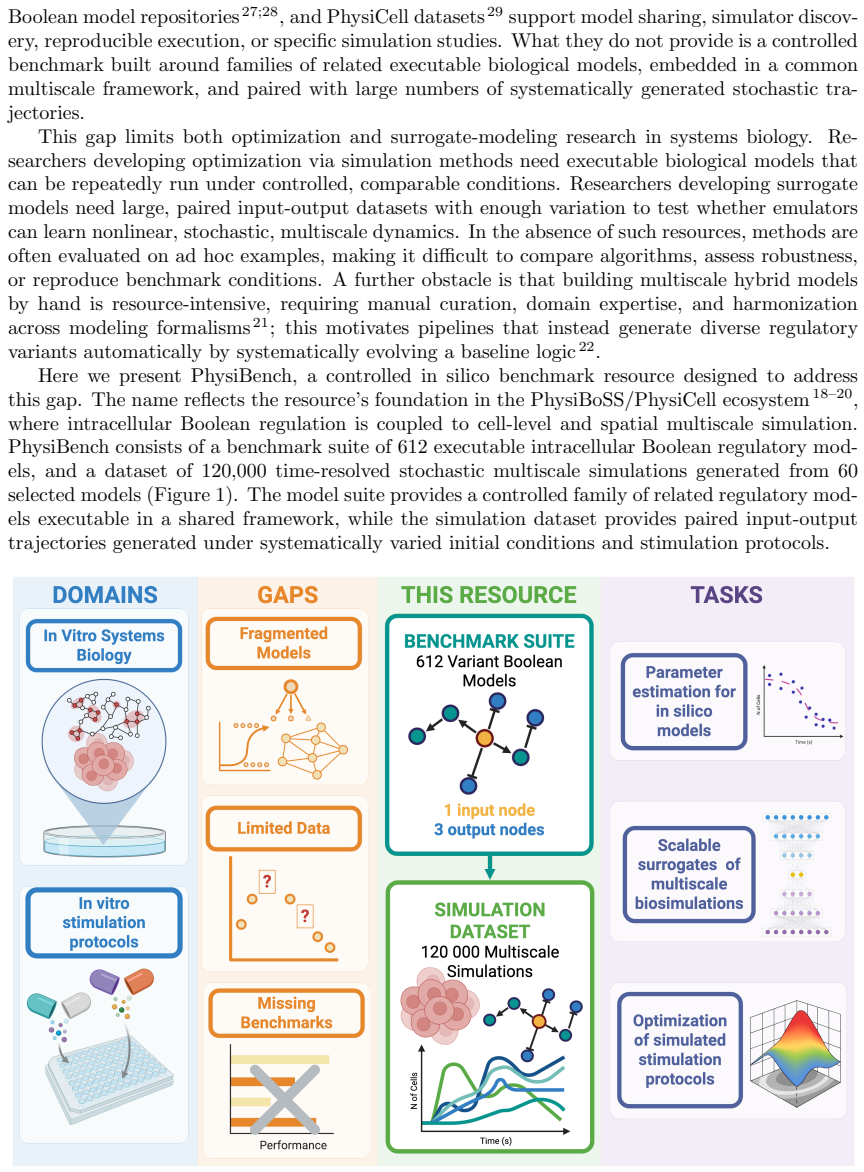
PhysiBench supplies 612 executable Boolean network variants and 120000 multiscale simulations as a benchmark for systems biology methods.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
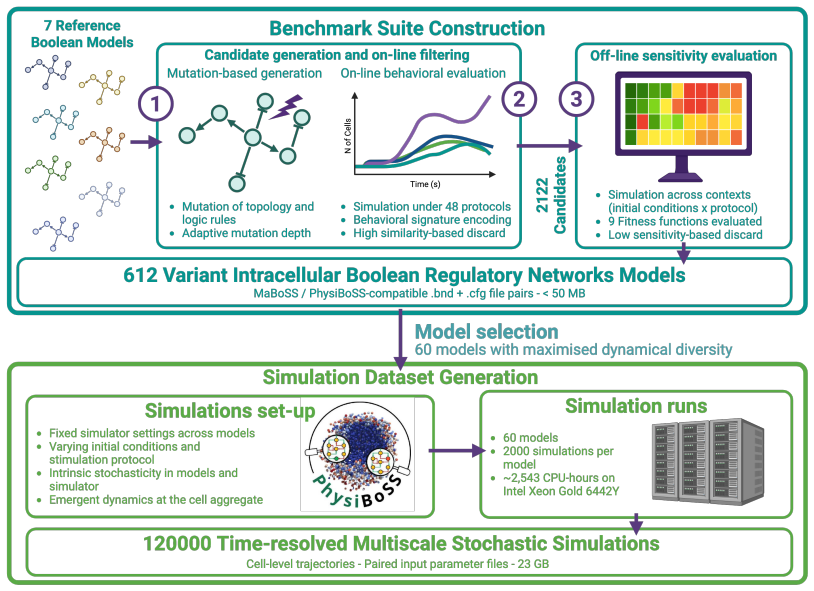
The central discovery is a benchmark suite of 612 executable Boolean model variants and 120000 linked multiscale simulation trajectories derived from seven source networks through mutation-based construction, behavioral filtering, and sensitivity evaluation, validated for structural diversity via graph analyses and for behavioral heterogeneity via multiscale outputs, enabling workflows including direct simulation in PhysiBoSS/PhysiCell, surrogate modeling, data-driven inference, simulation-based optimization, and comparative benchmarking.
What carries the argument
Mutation-based model construction combined with online behavioral filtering and offline sensitivity evaluation applied to seven published Boolean networks to generate executable variants.
If this is right
- The models can be executed directly under systematically sampled stimulation protocols with fixed initial configurations in the PhysiBoSS/PhysiCell framework.
- The linked dataset of 120000 trajectories enables development and testing of surrogate modeling and data-driven inference methods.
- Comparative benchmarking of different computational methods is facilitated by the standardized model identifiers and output files.
- Graph-based structural diversity analyses and behavioral heterogeneity assessments from multiscale outputs provide quantitative validation of the suite.
- The resource supports simulation-based optimization tasks across cell-cycle control, developmental patterning, cancer signaling, immune response, and cell-fate decision networks.
Where Pith is reading between the lines
- The public availability of linked model and simulation files could reduce duplicated effort when researchers build test cases for new algorithms.
- Community extensions might add further source networks or vary the simulation framework while retaining the same validation pipeline.
- The format of model identifiers plus input-parameter and output files could be adopted as a de-facto standard for sharing multiscale Boolean simulation data.
Load-bearing premise
The mutation-based construction, filtering, and sensitivity steps applied to the seven source networks produce variants that are sufficiently diverse, executable, and representative to serve as an effective benchmark suite.
What would settle it
A finding that a substantial fraction of the 612 models fail executability checks or that graph analyses show low structural diversity and simulation outputs lack measurable behavioral heterogeneity would undermine the claim that the suite functions as a useful benchmark.
Figures

read the original abstract
We present PhysiBench, an open resource for developing and evaluating computational methods in systems biology including a benchmark suite of 612 executable intracellular Boolean regulatory network variants and a dataset of 120,000 time-resolved multiscale stochastic simulations. The benchmark models are derived from seven published Boolean networks spanning cell-cycle control, developmental patterning, cancer signaling, immune response, and cell-fate decisions, and are executable in the PhysiBoSS/PhysiCell multiscale simulation framework. Model variants are generated through mutation-based model construction, online behavioral filtering, and offline sensitivity evaluation. The simulation dataset is produced from 60 selected models under systematically sampled stimulation protocols and fixed model-level initial configurations. Each trajectory is linked to its model identifier, input-parameter file, stochastic seed, and cell-level output file. PhysiBench supports direct simulation, surrogate modeling, data-driven inference, simulation-based optimization, and comparative benchmarking. Technical validation includes file-integrity and executability checks, graph-based structural diversity analyses, and behavioral heterogeneity assessment from multiscale simulation outputs.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper presents PhysiBench, an open resource consisting of a benchmark suite of 612 executable intracellular Boolean regulatory network variants derived from seven published networks (spanning cell-cycle, developmental, cancer, immune, and cell-fate processes) via mutation-based construction, online behavioral filtering, and offline sensitivity evaluation, together with a linked dataset of 120,000 time-resolved multiscale stochastic simulations produced from 60 selected models under sampled stimulation protocols. The models are executable in PhysiBoSS/PhysiCell; trajectories are linked to model identifiers, input files, seeds, and outputs. Technical validation comprises file-integrity and executability checks, graph-based structural diversity analyses, and behavioral heterogeneity assessment from simulation outputs. The resource is positioned to support direct simulation, surrogate modeling, data-driven inference, simulation-based optimization, and comparative benchmarking.
Significance. If the validation steps demonstrably establish sufficient structural and behavioral diversity together with executability, PhysiBench would constitute a useful standardized, open benchmark resource for computational systems biology. The explicit linkage of each trajectory to its originating model, parameters, and stochastic seed, combined with the multiscale simulation framework, would facilitate reproducible method development and comparative evaluation in an area that currently lacks such curated suites.
major comments (1)
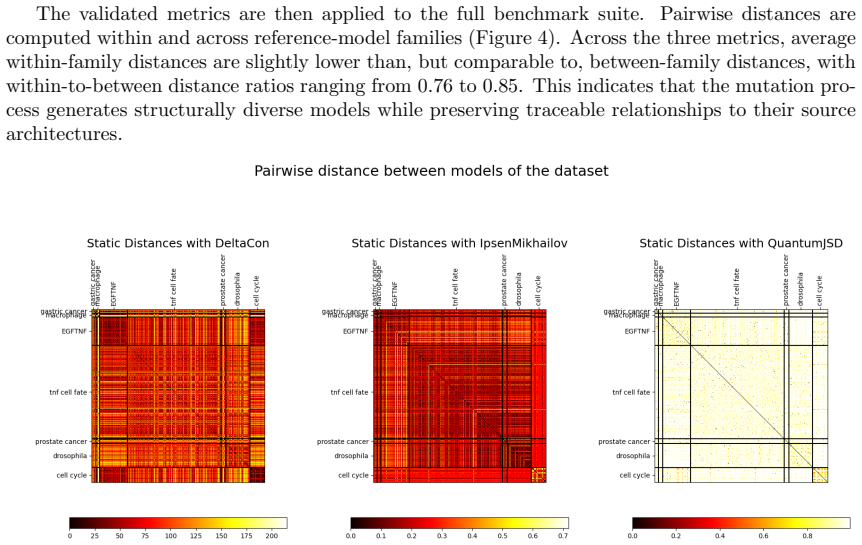
- [Abstract] Abstract: the technical validation is described only at a high level (file-integrity checks, graph-based structural diversity analyses, behavioral heterogeneity assessment) with no quantitative results, specific metrics (e.g., ranges of graph distances or heterogeneity scores), or exclusion criteria reported. This information is load-bearing for the central claim that the 612 variants plus 120k trajectories form a usable, validated benchmark suite.
minor comments (3)
- A summary table listing the seven source networks, the number of variants generated per network, and the final counts after filtering would improve clarity and allow readers to assess coverage.
- The selection process for the 60 models used to generate the 120k trajectories should be stated explicitly (e.g., stratification criteria or sensitivity thresholds).
- All original publications for the seven source Boolean networks must be cited with full references.
Simulated Author's Rebuttal
We thank the referee for the positive assessment of PhysiBench and the constructive comment on the abstract. We address the point below.
read point-by-point responses
-
Referee: [Abstract] Abstract: the technical validation is described only at a high level (file-integrity checks, graph-based structural diversity analyses, behavioral heterogeneity assessment) with no quantitative results, specific metrics (e.g., ranges of graph distances or heterogeneity scores), or exclusion criteria reported. This information is load-bearing for the central claim that the 612 variants plus 120k trajectories form a usable, validated benchmark suite.
Authors: We agree that the abstract describes the technical validation at a high level without quantitative metrics or explicit exclusion criteria. The full manuscript reports these details in the Technical Validation section, including graph-based structural analyses and behavioral heterogeneity from the multiscale outputs. To address the concern, we will revise the abstract to include representative quantitative results (e.g., observed ranges for structural diversity metrics and heterogeneity scores) and note the key filtering criteria used to select the final set of 612 variants. This change will make the abstract self-contained while preserving its length constraints. revision: yes
Circularity Check
No significant circularity
full rationale
The paper constructs and releases an explicit benchmark resource (612 model variants + 120k trajectories) via a described pipeline of mutation-based generation, online filtering, and offline sensitivity evaluation, followed by matching technical validation steps (file integrity, graph metrics, behavioral heterogeneity). No equations, fitted parameters, or first-principles predictions are claimed whose outputs reduce by construction to the inputs; the central claim is simply the existence and executability of the constructed suite, which is supported directly by the construction and validation process without self-referential reduction or load-bearing self-citation chains.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Cascarano, A., Mur-Petit, J., Hernandez-Gonzalez, J., Camacho, M., de Toro Eadie, N., Gkontra, P., Chadeau-Hyam, M., Vitria, J., and Lekadir, K. Machine and deep learning for longitudinal biomedical data: a review of methods and applications.Artificial Intelligence Review, 56(Suppl 2):1711–1771 (2023). https://doi.org/10.1007/s10462-023-10561-w
-
[2]
Mgod: Multi-granular outlier detection with 44 clustlier analysis
Abrate, M. P., Smeriglio, R., Bardini, R., Savino, A., and Di Carlo, S. Fast and accu- rate LSTM meta-modeling of TNF-induced tumor resistancein vitro. InProceedings of the 2024 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), 6194–6201 (2024). https://doi.org/10.1109/BIBM62325.2024.10822769
-
[3]
Advances in surrogate modeling for biological agent-based simulations: trends, challenges, and future prospects,
K. A. Norton, D. Bergman, H. V. Jain, and T. Jackson, “Advances in surrogate modeling for biological agent-based simulations: trends, challenges, and future prospects,”Journal of Mathematical Biology, vol. 92, no. 1, p. 6, 2026
2026
-
[4]
Ponce-de-Le´ on, M., Montagud, A., Akasiadis, C., Schreiber, J., Ntiniakou, T., and Valencia, A. Optimizing dosage-specific treatments in a multiscale model of tumor growth.Frontiers in Molecular Biosciences, 9:836794 (2022). https://doi.org/10.3389/fmolb.2022.836794
-
[5]
Bardini, R. and Di Carlo, S. Computational methods for biofabrication in tissue engineering and regenerative medicine—a literature review.Computational and Structural Biotechnology Journal, 23:601–616 (2024). https://doi.org/10.1016/j.csbj.2023.12.035
-
[6]
Castrignano, A., Bardini, R., Savino, A., and Di Carlo, S. A methodology combining rein- forcement learning and simulation to optimize the in silico culture of epithelial sheets.Journal of Computational Science, 76:102226 (2024). https://doi.org/10.1016/j.jocs.2024.102226
-
[7]
A methodology for co-simulation- based optimization of biofabrication protocols
Giannantoni, L., Bardini, R., and Di Carlo, S. A methodology for co-simulation- based optimization of biofabrication protocols. InProceedings of the International Work- Conference on Bioinformatics and Biomedical Engineering, pp. 179–192 (2022). Springer. https://doi.org/10.1007/978-3-031-07802-6 17
-
[8]
& Niepert, M.PDEBench: An Extensive Benchmark for Scientific Machine Learning
Takamoto, M., Praditia, T., Leiteritz, R., MacKinlay, D., Alesiani, F., Pfl¨ uger, D. & Niepert, M.PDEBench: An Extensive Benchmark for Scientific Machine Learning. In: Advances in Neural Information Processing Systems 36 (NeurIPS 2022), Datasets and Bench- marks Track. Available at:https://papers.neurips.cc/paper_files/paper/2022/file/ 0a9747136d411fb83f...
2022
-
[9]
GitHub repository
PDEBench Project.PDEBench: An Extensive Benchmark for Scientific Machine Learning. GitHub repository. Available at:https://github.com/pdebench/PDEBenchAccessed: 24 March 2026
2026
-
[10]
& Niepert, M.PDEBench Datasets
Takamoto, M., Praditia, T., Leiteritz, R., MacKinlay, D., Alesiani, F., Pfl¨ uger, D. & Niepert, M.PDEBench Datasets. DaRUS dataset. doi:10.18419/DARUS-2986. Available at:https:// darus.uni-stuttgart.de/dataset.xhtml?persistentId=doi:10.18419/darus-2986Ac- cessed: 24 March 2026
-
[11]
and Scher, Sebastian and Weyn, Jonathan A
Rasp, S., D¨ uben, P. D., Scher, S., Weyn, J. A., Mouatadid, S. & Thuerey, N.Weather- Bench: A Benchmark Data Set for Data-Driven Weather Forecasting.Journal of Advances in Modeling Earth Systems12, e2020MS002203 (2020). doi:10.1029/2020MS002203. Available at:https://agupubs.onlinelibrary.wiley.com/doi/10.1029/2020MS002203Accessed: 24 March 2026
-
[12]
GitHub repository
Pangeo Data.WeatherBench: A benchmark dataset for data-driven weather forecasting. GitHub repository. Available at:https://github.com/pangeo-data/WeatherBenchAc- cessed: 24 March 2026. 17
2026
-
[13]
GitHub repository
Google Research.WeatherBench 2: A benchmark for the next generation of data-driven global weather models. GitHub repository. Available at:https://github.com/google-research/ weatherbench2Accessed: 24 March 2026
2026
-
[14]
The Well: a Large-Scale Collection of Diverse Physics Simulations for Machine Learning.Advances in Neural Information Processing Systems37, 44989–45037 (2024)
Ohana, R.et al. The Well: a Large-Scale Collection of Diverse Physics Simulations for Machine Learning.Advances in Neural Information Processing Systems37, 44989–45037 (2024). Available at:https://openreview.net/forum?id=00Sx577BT3Accessed: 24 March 2026
2024
-
[15]
GitHub repository
Polymathic AI.The Well: 15TB of Physics Simulations. GitHub repository. Available at: https://github.com/PolymathicAI/the_wellAccessed: 24 March 2026
2026
-
[16]
Alsalloum, G. A., Al Sawaftah, N. M., Percival, K. M., and Husseini, G. A. Digital twins of biological systems: A narrative review.IEEE Open Journal of Engineering in Medicine and Biology(2024). https://doi.org/10.1109/OJEMB.2024.3426916
-
[17]
Bardini, R., Politano, G., Benso, A., and Di Carlo, S. Multi-level and hybrid modeling ap- proaches for systems biology.Computational and Structural Biotechnology Journal, 15:396–402 (2017). https://doi.org/10.1016/j.csbj.2017.05.001
-
[18]
https://doi.org/10.1093/bioinformatics/btx123
Stoll, G., Caron, B., Viara, E., Dugourd, A., Zinovyev, A., Naldi, A.,et al.MaBoSS 2.0: an environment for stochastic Boolean modeling.Bioinformatics, 33(14):2226–2228 (2017). https://doi.org/10.1093/bioinformatics/btx123
-
[19]
Letort, G., Montagud, A., Stoll, G., Heiland, R., Barillot, E., Macklin, P.,et al., and Calzone, L. PhysiBoSS: a multiscale agent-based modeling framework inte- grating physical dimension and cell signalling.Bioinformatics, 35(7):1188–1196 (2019). https://doi.org/10.1093/bioinformatics/bty766
-
[20]
Calzone, L., No¨ el, V., Barillot, E., Kroemer, G., and Stoll, G. modeling signaling pathways in biology with MaBoSS: From one single cell to a dynamic population of heterogeneous interacting cells.Computational and Structural Biotechnology Journal, 20:5661–5671 (2022). https://doi.org/10.1016/j.csbj.2022.09.034
-
[21]
Ruscone, M., Tsirvouli, E., Checcoli, A., Turei, D., Barillot, E., Saez-Rodriguez, J.,et al. NeKo: a tool for automatic network construction from prior knowledge.PLOS Computational Biology, 21(9):e1013300 (2025). https://doi.org/10.1371/journal.pcbi.1013300
-
[22]
Ouellet, M., Kim, J. Z., Guillaume, H., Shaffer, S. M., Bassett, L. C., and Bassett, D. S. Break- ing reflection symmetry: evolving long dynamical cycles in Boolean systems.New Journal of Physics, 26(2):023006 (2024). https://doi.org/10.1088/1367-2630/ad1b5a
-
[23]
Malik-Sheriff, R. S., Glont, M., Nguyen, T. V., Tiwari, K., Roberts, M. G., Xavier, A.,et al., and Hermjakob, H. BioModels—15 years of sharing computational models in life science. Nucleic Acids Research, 48(D1):D407–D415 (2020). https://doi.org/10.1093/nar/gkz1055
-
[24]
From quantitative SBML models to Boolean networks.Applied Network Science, 7(1):73 (2022)
Vaginay, A., Boukhobza, T., and Sma¨ ıl-Tabbone, M. From quantitative SBML models to Boolean networks.Applied Network Science, 7(1):73 (2022). https://doi.org/10.1007/s41109- 022-00473-8
-
[25]
P., Vasilescu, D., Marupilla, G., Wilson, M., Agmon, E., Agnew, H., Andrews, S
Shaikh, B., Smith, L. P., Vasilescu, D., Marupilla, G., Wilson, M., Agmon, E., Agnew, H., Andrews, S. S., Anwar, A., Beber, M. E., et al. BioSimulators: a central registry of simulation engines and services for recommending specific tools.Nucleic Acids Research, 50(W1):W108– W114 (2022). https://doi.org/10.1093/nar/gkac231
-
[26]
PhysiBoSS-Models: A database for multiscale models.arXiv preprint arXiv:2508.05550(2025)
Noel, V., Ruscone, M., Heiland, R., Montagud, A., Valencia, A., Barillot, E., Macklin, P., and Calzone, L. PhysiBoSS-Models: A database for multiscale models.arXiv preprint arXiv:2508.05550(2025). https://arxiv.org/abs/2508.05550
arXiv 2025
-
[27]
& Henzinger, T.Repository of logically consis- tent real-world Boolean network models
Pastva, S., ˇSafr´ anek, D., Beneˇ s, N., Brim, L. & Henzinger, T.Repository of logically consis- tent real-world Boolean network models. bioRxiv. Available at:https://www.biorxiv.org/ content/10.1101/2023.06.12.544361v1.fullAccessed: 24 March 2026. 18
work page doi:10.1101/2023.06.12.544361v1.fullaccessed: 2023
-
[28]
GitHub repository
Sybila.Biodivine Boolean Models (BBM) Benchmark Dataset. GitHub repository. Available at:https://github.com/sybila/biodivine-boolean-modelsAccessed: 24 March 2026
2026
-
[29]
L.Parameter Sensitivity in Tumor Models: Dataset from PhysiCell Simulations
Rocha, H. L.Parameter Sensitivity in Tumor Models: Dataset from PhysiCell Simulations. Zenodo dataset. doi:10.5281/zenodo.14590312. Available at:https://zenodo.org/records/ 14590312Accessed: 24 March 2026
-
[30]
Faur´ e, A., Naldi, A., Chaouiya, C., and Thieffry, D. Dynamical analysis of a generic Boolean model for the control of the mammalian cell cycle.Bioinformatics22(14), e124–e131 (2006). https://doi.org/10.1093/bioinformatics/btl210
-
[31]
Gene Interaction Network simulation (GINsim) Model Repository
Faur´ e, A.Restriction point control of the mammalian cell cycle. Gene Interaction Network simulation (GINsim) Model Repository. Available at:https://ginsim.github.io/models/ 2006-mammal-cell-cycle/Accessed: 23 March 2026
2006
-
[32]
and Othmer, H
Albert, R. and Othmer, H. G. The topology of the regulatory interactions predicts the expres- sion pattern of the segment polarity genes inDrosophila melanogaster.Journal of Theoretical Biology223(1), 1–18 (2003)
2003
-
[33]
Stabilizing patterning in the Drosophilasegment polarity network by selecting modelsin silico.Biosystems102(1), 3–10 (2010)
Stoll, G., Bischofberger, M., Rougemont, J., and Naef, F. Stabilizing patterning in the Drosophilasegment polarity network by selecting modelsin silico.Biosystems102(1), 3–10 (2010)
2010
-
[34]
MaBoSS model repository
MaBoSS Project.Drosophila Patterning. MaBoSS model repository. Available at:https: //maboss.curie.fr/Accessed: 23 March 2026
2026
-
[35]
M., Naldi, A., Van Iersel, M
Chaouiya, C., B´ erenguier, D., Keating, S. M., Naldi, A., Van Iersel, M. P., Rodriguez, N., et al.SBML qualitative models: a model representation format and infrastructure to foster interactions between qualitative modeling formalisms and tools.BMC Systems Biology7(1), 135 (2013)
2013
-
[36]
BioMod- els Database, BIOMD0000000562
BioModels.Chaouiya2013 - EGF and TNFalpha mediated signalling pathway. BioMod- els Database, BIOMD0000000562. Available at:https://www.ebi.ac.uk/biomodels/ BIOMD0000000562Accessed: 23 March 2026
2026
-
[37]
Discovery of drug synergies in gastric cancer cells predicted by logical modeling.PLoS Computational Biology11(8), e1004426 (2015)
Flobak, ˚A., Baudot, A., Remy, E., Thommesen, L., Thieffry, D., Kuiper, M., and Lægreid, A. Discovery of drug synergies in gastric cancer cells predicted by logical modeling.PLoS Computational Biology11(8), e1004426 (2015)
2015
-
[38]
PhysiBoSS boolean-models repository
PhysiBoSS.gastric cancer. PhysiBoSS boolean-models repository. Available at:https:// github.com/PhysiBoSS/boolean-models/tree/main/gastric_cancerAccessed: 23 March 2026
2026
-
[39]
E.,et al., and the COVID-19 Disease Map Community
Niarakis, A., Ostaszewski, M., Mazein, A., Kuperstein, I., Kutmon, M., Gillespie, M. E.,et al., and the COVID-19 Disease Map Community. Drug-target identification in COVID-19 dis- ease mechanisms using computational systems biology approaches.Frontiers in Immunology, 14:1282859 (2024). https://doi.org/10.3389/fimmu.2023.1282859
-
[40]
pb4covid19 repository
PhysiBoSS-COVID.boolean network. pb4covid19 repository. Available at: https://gitlab.lcsb.uni.lu/computational-modeling-and-simulation/pb4covid19/ -/tree/master/data/boolean_networkAccessed: 23 March 2026
2026
-
[41]
Montagud, A., B´ eal, J., Tobalina, L., Traynard, P., Subramanian, V., Szalai, B.,et al.Patient- specific Boolean models of signalling networks guide personalised treatments.eLife11, e72626 (2022)
2022
-
[42]
PhysiBoSS boolean-models repository
PhysiBoSS.prostate cancer. PhysiBoSS boolean-models repository. Available at:https: //github.com/PhysiBoSS/boolean-models/tree/main/prostate_cancerAccessed: 23 March 2026. 19
2026
-
[43]
Mathematical modeling of cell-fate decision in response to death receptor engage- ment.PLoS Computational Biology6(3), e1000702 (2010)
Calzone, L., Tournier, L., Fourquet, S., Thieffry, D., Zhivotovsky, B., Barillot, E., and Zi- novyev, A. Mathematical modeling of cell-fate decision in response to death receptor engage- ment.PLoS Computational Biology6(3), e1000702 (2010)
2010
-
[44]
PhysiBoSS boolean-models repository
PhysiBoSS.tnf cell fate. PhysiBoSS boolean-models repository. Available at:https:// github.com/PhysiBoSS/boolean-models/tree/main/tnf_cell_fateAccessed: 23 March 2026
2026
-
[45]
Benesty, J., Chen, J., Huang, Y., and Cohen, I. Pearson correlation coefficient. InNoise Reduction in Speech Processing, pp. 1–4 (2009). Springer. https://doi.org/10.1007/978-3-642- 00296-0 5
-
[46]
McCabe, S., Torres, L., LaRock, T., Haque, S. A., Yang, C. H., Hartle, H., and Klein, B.netrd: A library for network reconstruction and graph distances.arXiv preprintarXiv:2010.16019 (2020)
arXiv 2010
-
[47]
J., and Schult, D
Hagberg, A., Swart, P. J., and Schult, D. A. Exploring network structure, dynamics, and function usingNetworkX. Los Alamos National Laboratory Technical Report LA-UR-08-05495 (2008)
2008
-
[48]
T., and Faloutsos, C
Koutra, D., Vogelstein, J. T., and Faloutsos, C. DeltaCon: A principled massive-graph simi- larity function. InProceedings of the 2013 SIAM International Conference on Data Mining, 162–170 (2013)
2013
-
[49]
Biological network comparison via Ipsen–Mikhailov distance
Jurman, G., Riccadonna, S., Visintainer, R., and Furlanello, C. Biological network comparison via Ipsen–Mikhailov distance. arXiv preprint arXiv:1109.0220 (2011)
Pith/arXiv arXiv 2011
-
[50]
Nielsen, F. On a variational definition for the Jensen–Shannon symmetriza- tion of distances based on the information radius.Entropy23(4), 464 (2021). https://doi.org/10.3390/e21050485
-
[51]
PhysiBench resource
Masera, M., Smeriglio, R., Bardini, R., Savino, A., Di Carlo, S.PhysiBench. PhysiBench resource. Available at:https://drive.cloud.polito.it/index.php/s/pefzfJpnZZiRMWY Accessed: 16 June 2026
2026
-
[52]
GitHub repository
PhysiBench GitHub repository.PhysiBench GitHub repository. GitHub repository. Available at:https://github.com/smilies-polito/PhysiBenchAccessed: 16 June 2026. 20
2026
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.