Surveying the adaptive landscapes of 10,000 antibodies
Pith reviewed 2026-06-26 12:42 UTC · model grok-4.3
The pith
A parameter-free framework using convergent mutations in public clonotypes identifies beneficial antibody mutations and a prevalence-fitness tradeoff across more than 10,000 examples.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
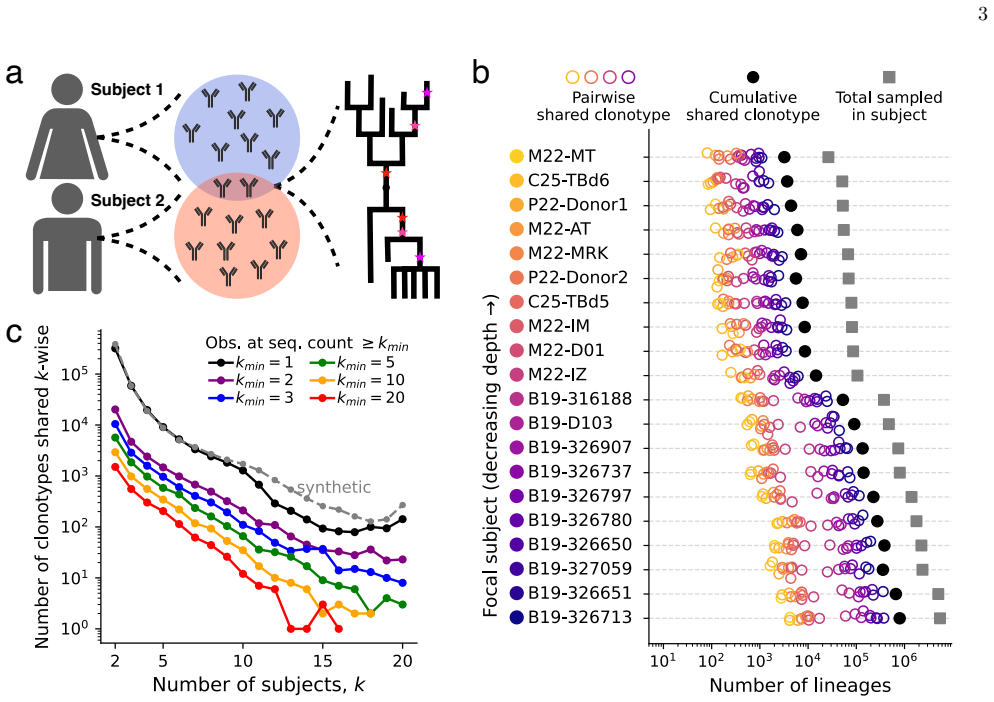
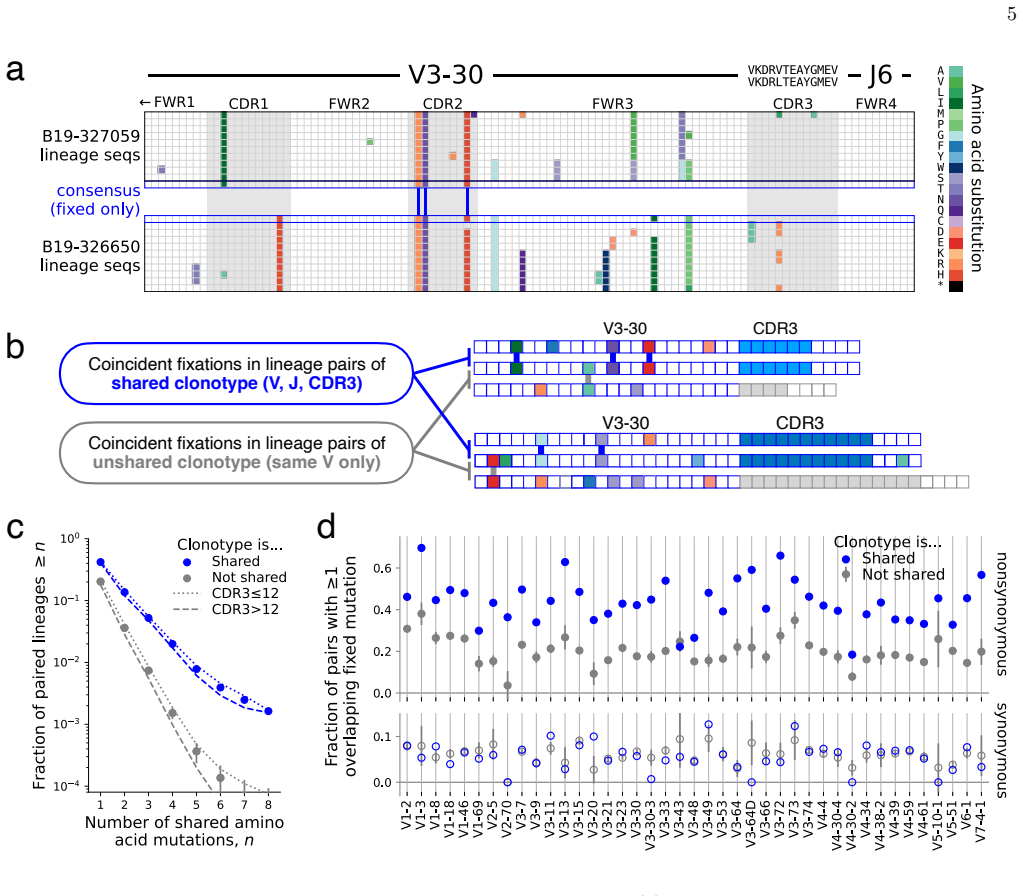
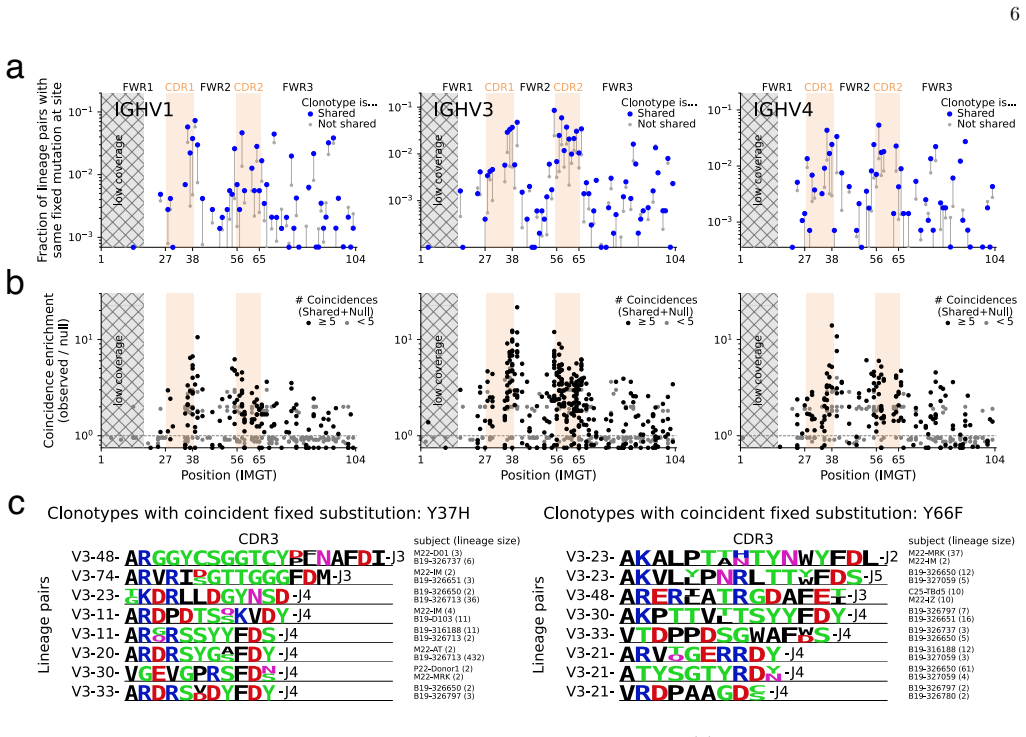
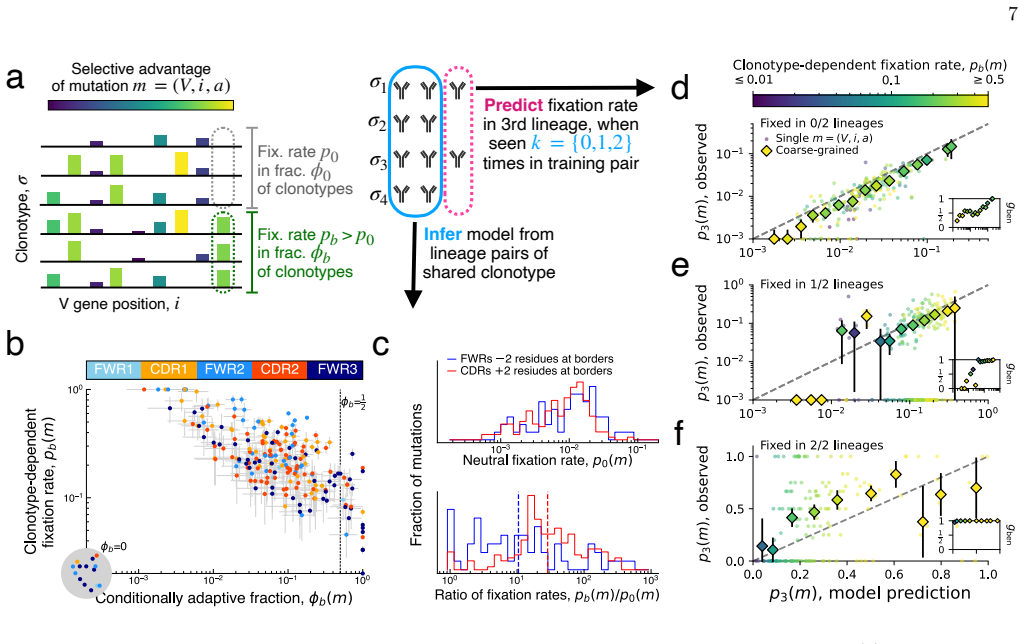
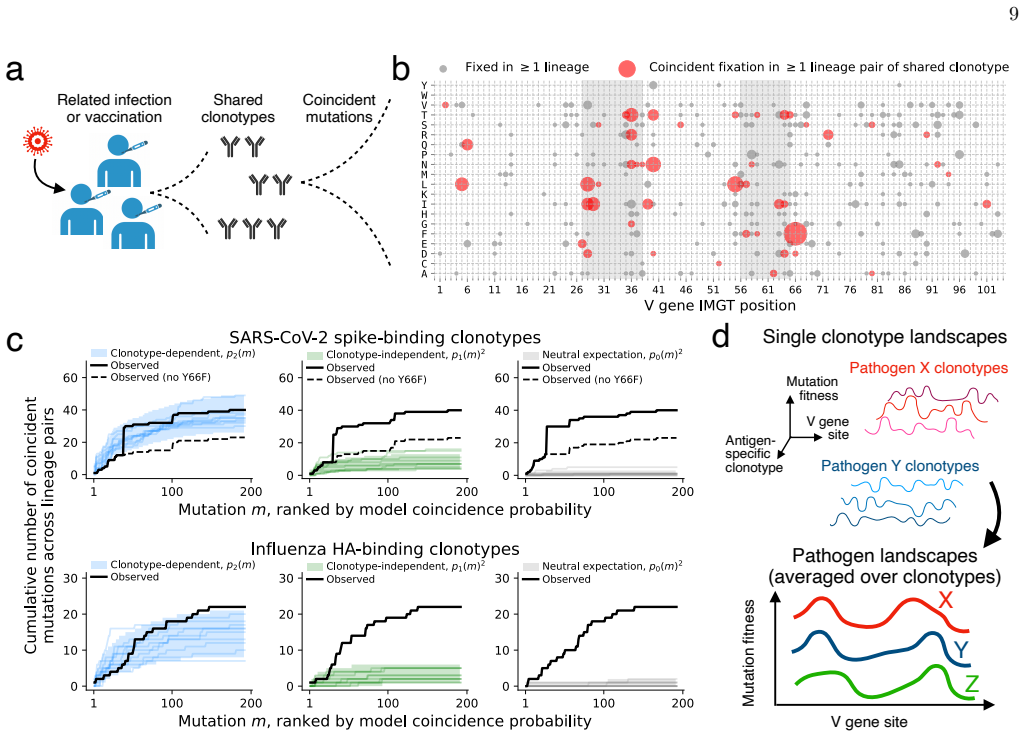
By applying a parameter-free population genetic framework to the statistics of convergent affinity maturation in more than 10,000 public clonotypes represented by multiple lineages across 20 healthy individuals, the authors identify widespread signatures of clonotype-dependent selection of individual mutations. They estimate the prevalence and typical fitness effects of mutations across the V gene at the single-site level, uncovering a general tradeoff between prevalence and fitness effect. These inferred landscapes broadly reproduce the statistics of convergent mutation in antibodies specific to SARS-CoV-2 and influenza. The framework also benchmarks predictions from existing antibody langu
What carries the argument
The parameter-free population genetic framework that leverages the statistics of convergent affinity maturation in public clonotypes (B cell lineages sharing similar naive sequences) to identify beneficial mutations.
If this is right
- Selection acts on mutations in a manner that depends on the specific clonotype rather than uniformly across all antibodies.
- A tradeoff exists such that mutations observed in more lineages tend to confer smaller fitness improvements.
- The inferred single-site landscapes reproduce observed convergent mutation frequencies in antibodies against specific pathogens.
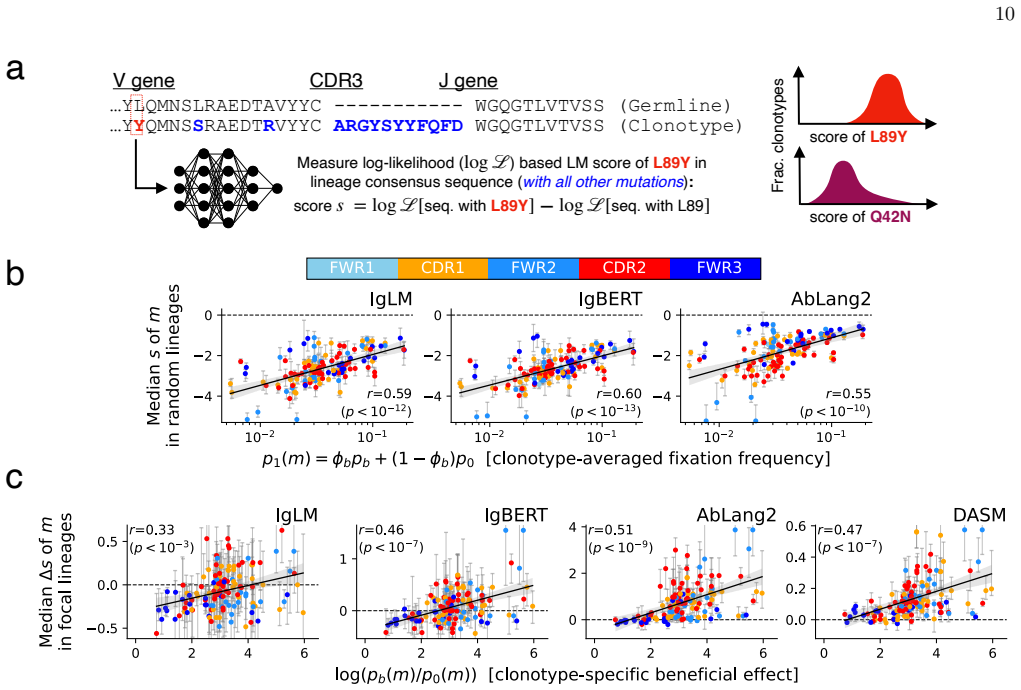
- Antibody language models primarily capture non-selective sequence patterns, but renormalization isolates the selection component.
Where Pith is reading between the lines
- Antibody design efforts could prioritize mutations predicted to be selected within a target clonotype rather than using averaged landscapes.
- The approach may generalize to other immune receptors or evolutionary systems that exhibit repeated changes from similar starting points.
- Collecting data from additional individuals would likely sharpen estimates of how fitness effects vary with mutation prevalence.
Load-bearing premise
The observed statistics of convergent mutations within public clonotypes directly reflect clonotype-dependent positive selection on individual sites, without confounding from shared naive sequence biases or sampling effects across individuals.
What would settle it
Observing that convergent mutation patterns within public clonotypes can be fully accounted for by properties of the naive sequences alone or by sampling variation, or that the inferred beneficial mutations fail to appear at higher rates in actual SARS-CoV-2 and influenza antibody responses.
Figures



read the original abstract
Affinity maturation is the Darwinian process by which antibodies improve antigen binding through somatic hypermutation and selection. The adaptive landscape, which defines the set of antibody-specific mutations that improve functional characteristics like antigen binding, has been explored in only a handful of antibodies. Identifying the sites of adaptive mutations in a given antibody sequence, and how these sites vary across the antibody repertoire, can inform the design of therapeutic antibodies. We develop a parameter-free population genetic framework that leverages the statistics of convergent affinity maturation in B cell lineages sharing similar naive sequences, called public clonotypes, to identify beneficial mutations. Applying this framework to more than 10,000 public clonotypes represented by multiple lineages across 20 healthy individuals, we identify widespread signatures of clonotype-dependent selection of individual mutations. We estimate the prevalence and typical fitness effects of mutations across the V gene at the single-site level, uncovering a general tradeoff between prevalence and fitness effect. These inferred landscapes broadly reproduce the statistics of convergent mutation in antibodies specific to SARS-CoV-2 and influenza. Finally, we use our framework to benchmark predictions from existing antibody language models, and show that while these models are dominated by non-selective signatures, a simple renormalization procedure can expose signatures of clonotype-dependent positive selection consistent with our predictions.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper develops a parameter-free population genetic framework that uses statistics of convergent affinity maturation in public clonotypes (B cell lineages sharing similar naive sequences) to infer site-specific beneficial mutations and adaptive landscapes. Applied to >10,000 public clonotypes across 20 healthy individuals, it reports widespread clonotype-dependent selection signatures, a general tradeoff between mutation prevalence and fitness effect, reproduction of convergent mutation statistics in SARS-CoV-2 and influenza antibodies, and a renormalization procedure to extract selection signals from antibody language models.
Significance. If the framework isolates selection without hidden parameters or post-hoc adjustments, the scale of the survey (10k+ clonotypes) and the reproduction of known convergent patterns would represent a substantial advance in mapping antibody adaptive landscapes, with direct relevance to therapeutic design. The explicit parameter-free claim and use for model benchmarking are positive features that could be cited if the central inference holds.
major comments (2)
- [Abstract] Abstract: The central inference attributes excess convergence of specific mutations within public clonotypes to clonotype-dependent positive selection. However, no explicit null model or correction is described for mutation-rate heterogeneity or sequence-context biases that could correlate with the naive V-gene similarity used to define clonotypes, which could produce the same patterns under neutrality.
- [Abstract] Abstract: The reported 'general tradeoff between prevalence and fitness effect' and the reproduction of convergent mutation statistics rest on unexamined data processing and statistical definitions; without verification that these quantities are extracted directly without fitted parameters or self-referential definitions, the load-bearing claims cannot be assessed.
minor comments (1)
- [Abstract] Abstract: The phrase 'broadly reproduce' lacks quantitative metrics (e.g., correlation coefficients or overlap statistics) that would clarify the strength of agreement with SARS-CoV-2 and influenza data.
Simulated Author's Rebuttal
We thank the referee for their detailed comments. We address each major point below with clarifications on the framework's controls and definitions. Our responses focus on the manuscript content without misrepresentation.
read point-by-point responses
-
Referee: The central inference attributes excess convergence of specific mutations within public clonotypes to clonotype-dependent positive selection. However, no explicit null model or correction is described for mutation-rate heterogeneity or sequence-context biases that could correlate with the naive V-gene similarity used to define clonotypes, which could produce the same patterns under neutrality.
Authors: The clonotype definition groups lineages by naive V-gene sequence similarity, ensuring comparable mutational contexts within each clonotype. The inference uses the statistic of repeated independent acquisition of the same mutation across lineages of one clonotype, which exceeds the baseline rate observed across clonotypes. This structure controls for context-dependent mutation rates because the starting sequences are matched; global biases uncorrelated with clonotype identity would not produce clonotype-specific convergence patterns. We will revise the abstract and add a methods paragraph explicitly describing this control and why separate null simulations are not required for the parameter-free claim. revision_made = partial revision: partial
-
Referee: The reported 'general tradeoff between prevalence and fitness effect' and the reproduction of convergent mutation statistics rest on unexamined data processing and statistical definitions; without verification that these quantities are extracted directly without fitted parameters or self-referential definitions, the load-bearing claims cannot be assessed.
Authors: Prevalence is the direct fraction of clonotypes containing the mutation at least once. The fitness effect is the within-clonotype convergence rate (fraction of lineages acquiring the mutation) normalized by the clonotype's overall mutation count, using raw counts with no fitted parameters. These definitions are independent: prevalence is a global count, while the convergence statistic is local to each clonotype. The SARS-CoV-2 and influenza reproductions apply identical count-based definitions to those datasets. We will add a methods subsection with explicit formulas and verification that no self-reference or fitting occurs. revision_made = yes revision: yes
Circularity Check
No circularity: parameter-free inference from empirical convergence counts
full rationale
The framework is explicitly parameter-free and derives site-wise prevalence and fitness-effect estimates directly from observed counts of convergent mutations within public clonotypes defined by naive-sequence similarity. No equations reduce a claimed prediction back to a fitted input by construction, no self-citation chain supplies the core uniqueness or ansatz, and the method does not rename a known empirical pattern under new coordinates. The derivation therefore remains self-contained against the input statistics; any concern about confounding by naive-sequence biases or sampling is a question of external validity rather than internal circularity.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption Convergent mutations across independent lineages sharing similar naive sequences indicate positive selection on those sites.
- domain assumption Public clonotypes across healthy individuals provide an unbiased sample for inferring general V-gene selection patterns.
Reference graph
Works this paper leans on
-
[1]
MacLennan IC (1994) Germinal centers.Annual review of immunology12:117–139
1994
-
[2]
Annual review of immunology30:429–457
Victora GD, Nussenzweig MC (2012) Germinal centers. Annual review of immunology30:429–457
2012
-
[3]
Annual review of immunology40:413–442
Victora GD, Nussenzweig MC (2022) Germinal centers. Annual review of immunology40:413–442
2022
-
[4]
(2025) Replaying germinal center evo- lution on a quantified affinity landscape.Cell
DeWitt WS, et al. (2025) Replaying germinal center evo- lution on a quantified affinity landscape.Cell
2025
-
[5]
Koenig P, et al. (2015) Deep sequencing-guided design of a high affinity dual specificity antibody to target two angiogenic factors in neovascular age-related macular de- generation.Journal of Biological Chemistry290:21773– 21786
2015
-
[6]
Adams RM, Mora T, Walczak AM, Kinney JB (2016) Measuring the sequence-affinity landscape of antibodies with massively parallel titration curves.Elife5:e23156
2016
-
[7]
Koenig P, et al. (2017) Mutational landscape of anti- body variable domains reveals a switch modulating the interdomain conformational dynamics and antigen bind- ing.Proceedings of the National Academy of Sciences 114:E486–E495
2017
-
[8]
Madan B, et al. (2021) Mutational fitness landscapes reveal genetic and structural improvement pathways for a vaccine-elicited hiv-1 broadly neutralizing anti- body.Proceedings of the National Academy of Sciences 118:e2011653118
2021
-
[9]
(2021) Binding affinity landscapes constrain the evolution of broadly neutralizing anti- influenza antibodies.Elife10:e71393
Phillips AM, et al. (2021) Binding affinity landscapes constrain the evolution of broadly neutralizing anti- influenza antibodies.Elife10:e71393
2021
-
[10]
(2022) Compensatory epistasis main- tains ace2 affinity in sars-cov-2 omicron ba
Moulana A, et al. (2022) Compensatory epistasis main- tains ace2 affinity in sars-cov-2 omicron ba. 1.Nature communications13:7011
2022
-
[11]
(2023) The landscape of antibody bind- ing affinity in sars-cov-2 omicron ba
Moulana A, et al. (2023) The landscape of antibody bind- ing affinity in sars-cov-2 omicron ba. 1 evolution.Elife 12:e83442
2023
-
[12]
Schulz S, Tan TJ, Wu NC, Wang S (2025) Epistatic hotspots organize antibody fitness landscape and boost evolvability.Proceedings of the National Academy of Sci- ences122:e2413884122
2025
-
[13]
(2025) Retrospective sars-cov-2 human antibody development trajectories are largely sparse and permissive.Proceedings of the National Academy of Sci- ences122:e2412787122
Kirby MB, et al. (2025) Retrospective sars-cov-2 human antibody development trajectories are largely sparse and permissive.Proceedings of the National Academy of Sci- ences122:e2412787122
2025
-
[14]
Nourmohammad A, Otwinowski J, Luksza M, Mora T, Walczak AM (2019) Fierce selection and interference in b-cell repertoire response to chronic hiv-1.Molecular bi- ology and evolution36:2184–2194
2019
-
[15]
Yaari G, Uduman M, Kleinstein SH (2012) Quantifying selection in high-throughput immunoglobulin sequencing data sets.Nucleic acids research40:e134–e134
2012
-
[16]
Yaari G, Benichou JI, Vander Heiden JA, Kleinstein SH, Louzoun Y (2015) The mutation patterns in b-cell im- munoglobulin receptors reflect the influence of selection acting at multiple time-scales.Philosophical Transactions of the Royal Society B: Biological Sciences370
2015
-
[17]
Horns F, Vollmers C, Dekker CL, Quake SR (2019) Signatures of selection in the human antibody reper- toire: Selective sweeps, competing subclones, and neutral drift.Proceedings of the National Academy of Sciences 116:1261–1266
2019
-
[18]
(2021) Human b cell lineages associated with germinal centers following influenza vaccination are measurably evolving.Elife10:e70873
Hoehn KB, et al. (2021) Human b cell lineages associated with germinal centers following influenza vaccination are measurably evolving.Elife10:e70873
2021
-
[19]
Ralph DK, Matsen IV FA (2020) Using b cell receptor lineage structures to predict affinity.PLOS Computa- tional Biology16:e1008391
2020
-
[20]
(2022) Memory persistence and differ- entiation into antibody-secreting cells accompanied by positive selection in longitudinal bcr repertoires.Elife 11:e79254
Mikelov A, et al. (2022) Memory persistence and differ- entiation into antibody-secreting cells accompanied by positive selection in longitudinal bcr repertoires.Elife 11:e79254
2022
- [21]
-
[22]
Bioinformatics Advances2:vbac046
Olsen TH, Moal IH, Deane CM (2022) Ablang: an anti- body language model for completing antibody sequences. Bioinformatics Advances2:vbac046
2022
-
[23]
Shuai RW, Ruffolo JA, Gray JJ (2023) Iglm: Infilling language modeling for antibody sequence design.Cell systems14:979–989
2023
-
[24]
(2024) Large scale paired anti- body language models.PLOS Computational Biology 20:e1012646
Kenlay H, et al. (2024) Large scale paired anti- body language models.PLOS Computational Biology 20:e1012646
2024
-
[25]
Burbach SM, Briney B (2024) Improving antibody lan- guage models with native pairing.Patterns5
2024
-
[26]
Burbach SM, Briney B (2025) A curriculum learning approach to training antibody language models.PLOS Computational Biology21:e1013473
2025
-
[27]
Olsen TH, Moal IH, Deane CM (2024) Addressing the an- tibody germline bias and its effect on language models for 15 improved antibody design.Bioinformatics40:btae618
2024
-
[28]
Ng K, Briney B (2025) Focused learning by anti- body language models using preferential masking of non- templated regions.Patterns6
2025
-
[29]
(2025) A sitewise model of natu- ral selection on individual antibodies via a transformer– encoder.Molecular Biology and Evolution42:msaf186
Matsen IV FA, et al. (2025) A sitewise model of natu- ral selection on individual antibodies via a transformer– encoder.Molecular Biology and Evolution42:msaf186
2025
-
[30]
(2026) Separating selection from mutation in antibody language models.Elife 15:RP109644
Matsen IV FA, et al. (2026) Separating selection from mutation in antibody language models.Elife 15:RP109644
2026
-
[31]
(2018) Multi-donor longitudinal antibody repertoire sequencing reveals the existence of public an- tibody clonotypes in hiv-1 infection.Cell host & microbe 23:845–854
Setliff I, et al. (2018) Multi-donor longitudinal antibody repertoire sequencing reveals the existence of public an- tibody clonotypes in hiv-1 infection.Cell host & microbe 23:845–854
2018
-
[32]
(2022) Sequence and functional char- acterization of a public hiv-specific antibody clonotype
Murji AA, et al. (2022) Sequence and functional char- acterization of a public hiv-specific antibody clonotype. Iscience25
2022
-
[33]
(2023) Convergent antibody responses are associated with broad neutralization of hepatitis c virus.Frontiers in immunology14:1135841
Skinner NE, et al. (2023) Convergent antibody responses are associated with broad neutralization of hepatitis c virus.Frontiers in immunology14:1135841
2023
-
[34]
(2019) Polyclonal and convergent an- tibody response to ebola virus vaccine rvsv-zebov.Nature medicine25:1589–1600
Ehrhardt SA, et al. (2019) Polyclonal and convergent an- tibody response to ebola virus vaccine rvsv-zebov.Nature medicine25:1589–1600
2019
-
[35]
(2021) Convergent antibody evolution and clonotype expansion following influenza virus vacci- nation.PLoS One16:e0247253
Forgacs D, et al. (2021) Convergent antibody evolution and clonotype expansion following influenza virus vacci- nation.PLoS One16:e0247253
2021
-
[36]
(2024) An explainable language model for antibody specificity prediction using curated influenza hemagglutinin antibodies.Immunity57:2453–2465
Wang Y, et al. (2024) An explainable language model for antibody specificity prediction using curated influenza hemagglutinin antibodies.Immunity57:2453–2465
2024
-
[37]
(2020) Convergent antibody re- sponses to sars-cov-2 in convalescent individuals.Nature 584:437–442
Robbiani DF, et al. (2020) Convergent antibody re- sponses to sars-cov-2 in convalescent individuals.Nature 584:437–442
2020
-
[38]
(2020) Deep sequencing of b cell re- ceptor repertoires from covid-19 patients reveals strong convergent immune signatures.Frontiers in immunology 11:605170
Galson JD, et al. (2020) Deep sequencing of b cell re- ceptor repertoires from covid-19 patients reveals strong convergent immune signatures.Frontiers in immunology 11:605170
2020
-
[39]
(2021) Convergent antibody responses to the sars-cov-2 spike protein in convalescent and vacci- nated individuals.Cell reports36
Chen EC, et al. (2021) Convergent antibody responses to the sars-cov-2 spike protein in convalescent and vacci- nated individuals.Cell reports36
2021
-
[40]
(2021) Sequence signatures of two public antibody clonotypes that bind sars-cov-2 receptor bind- ing domain.Nature Communications12:3815
Tan TJ, et al. (2021) Sequence signatures of two public antibody clonotypes that bind sars-cov-2 receptor bind- ing domain.Nature Communications12:3815
2021
-
[41]
(2022) A large-scale systematic survey reveals recurring molecular features of public antibody responses to sars-cov-2.Immunity55:1105–1117
Wang Y, et al. (2022) A large-scale systematic survey reveals recurring molecular features of public antibody responses to sars-cov-2.Immunity55:1105–1117
2022
-
[42]
(2022) The prominent role of a cdr1 somatic hypermutation for convergent ighv3-53/3-66 antibodies in binding to sars-cov-2.Emerging Microbes & Infections 11:1186–1190
Tian X, et al. (2022) The prominent role of a cdr1 somatic hypermutation for convergent ighv3-53/3-66 antibodies in binding to sars-cov-2.Emerging Microbes & Infections 11:1186–1190
2022
-
[43]
(2025) Clonotype-enriched somatic hyper- mutations drive affinity maturation of a public human an- tibody targeting an occluded sarbecovirus epitope.Cell reports44
Rao VN, et al. (2025) Clonotype-enriched somatic hyper- mutations drive affinity maturation of a public human an- tibody targeting an occluded sarbecovirus epitope.Cell reports44
2025
-
[44]
(2025) Ai identifies broadly neutralizing an- tibodies from an ighv1-69 public antibody class exerting continued selection over sars-cov-2.bioRxivpp 2025–12
Niu C, et al. (2025) Ai identifies broadly neutralizing an- tibodies from an ighv1-69 public antibody class exerting continued selection over sars-cov-2.bioRxivpp 2025–12
2025
-
[45]
Briney B, Inderbitzin A, Joyce C, Burton DR (2019) Commonality despite exceptional diversity in the baseline human antibody repertoire.Nature566:393–397
2019
-
[46]
(2022) Clonal structure, stability and dynamics of human memory b cells and circulating plas- mablasts.Nature immunology23:1076–1085
Phad GE, et al. (2022) Clonal structure, stability and dynamics of human memory b cells and circulating plas- mablasts.Nature immunology23:1076–1085
2022
-
[47]
Cvijovi´ c I, Swift M, Quake SR (2025) Long-term b cell memory emerges at uniform relative rates in the human immune response.Proceedings of the National Academy of Sciences122:e2406474122
2025
-
[48]
Spisak N, Ath` enes G, Dupic T, Mora T, Walczak AM (2024) Combining mutation and recombination statis- tics to infer clonal families in antibody repertoires.Elife 13:e86181
2024
-
[49]
Ruiz Ortega M, Spisak N, Mora T, Walczak AM (2023) Modeling and predicting the overlap of b-and t-cell re- ceptor repertoires in healthy and sars-cov-2 infected in- dividuals.PLoS Genetics19:e1010652
2023
-
[50]
(2015) Inferring processes underlying b-cell repertoire diversity.Philosophical Transactions of the Royal Society B: Biological Sciences370
Elhanati Y, et al. (2015) Inferring processes underlying b-cell repertoire diversity.Philosophical Transactions of the Royal Society B: Biological Sciences370
2015
-
[51]
Marcou Q, Mora T, Walczak AM (2018) High- throughput immune repertoire analysis with igor.Nature communications9:561
2018
-
[52]
Sethna Z, Elhanati Y, Callan Jr CG, Walczak AM, Mora T (2019) Olga: fast computation of generation proba- bilities of b-and t-cell receptor amino acid sequences and motifs.Bioinformatics35:2974–2981
2019
-
[53]
Isacchini G, Walczak AM, Mora T, Nourmohammad A (2021) Deep generative selection models of t and b cell receptor repertoires with sonnia.Proceedings of the Na- tional Academy of Sciences118:e2023141118
2021
-
[54]
Desponds J, Mora T, Walczak AM (2016) Fluctuating fit- ness shapes the clone-size distribution of immune reper- toires.Proceedings of the National Academy of Sciences 113:274–279
2016
-
[55]
arXiv preprint arXiv:2510.02812
Mazzolini A, Walczak AM, Mora T (2025) Dynamics of memory b cells and plasmablasts in healthy individuals. arXiv preprint arXiv:2510.02812
-
[56]
(1989) Conformations of immunoglob- ulin hypervariable regions.Nature342:877–883
Chothia C, et al. (1989) Conformations of immunoglob- ulin hypervariable regions.Nature342:877–883
1989
-
[57]
(2015) Quantifying evolutionary constraints on b-cell affinity maturation.Philosophical Transactions of the Royal Society B: Biological Sciences 370
McCoy CO, et al. (2015) Quantifying evolutionary constraints on b-cell affinity maturation.Philosophical Transactions of the Royal Society B: Biological Sciences 370
2015
-
[58]
(2022) Broadly neutralizing anti- bodies target a haemagglutinin anchor epitope.Nature 602:314–320
Guthmiller JJ, et al. (2022) Broadly neutralizing anti- bodies target a haemagglutinin anchor epitope.Nature 602:314–320
2022
-
[59]
Raju N, et al. (2024) Multiplexed antibody sequencing and profiling of the human hemagglutinin-specific mem- ory b cell response following influenza vaccination.The Journal of Immunology213:1605–1619
2024
-
[60]
(2026) B cell imprinting in children impairs antibodies to the haemagglutinin stalk.Naturepp 1–10
Sun J, et al. (2026) B cell imprinting in children impairs antibodies to the haemagglutinin stalk.Naturepp 1–10
2026
-
[61]
(2020) Structural basis of a shared anti- body response to sars-cov-2.Science369:1119–1123
Yuan M, et al. (2020) Structural basis of a shared anti- body response to sars-cov-2.Science369:1119–1123
2020
-
[62]
Chungyoun M, Gray J (2025) Fitness landscape for an- tibodies 2: Benchmarking reveals that protein ai models cannot yet consistently predict developability properties. bioRxiv
2025
-
[63]
Nijkamp E, Ruffolo JA, Weinstein EN, Naik N, Madani A (2023) Progen2: exploring the boundaries of protein language models.Cell systems14:968–978
2023
-
[64]
(2023) Evolutionary-scale prediction of atomic-level protein structure with a language model
Lin Z, et al. (2023) Evolutionary-scale prediction of atomic-level protein structure with a language model. Science379:1123–1130
2023
-
[65]
(2026) Scaling unlocks broader generation and deeper functional understanding of pro- teins.Advances in Neural Information Processing Sys- tems38:46109–46145
Bhatnagar A, et al. (2026) Scaling unlocks broader generation and deeper functional understanding of pro- teins.Advances in Neural Information Processing Sys- tems38:46109–46145
2026
-
[66]
Protein Science31:141–146
Olsen TH, Boyles F, Deane CM (2022) Observed anti- body space: A diverse database of cleaned, annotated, 16 and translated unpaired and paired antibody sequences. Protein Science31:141–146
2022
-
[67]
(2021) Language models enable zero-shot prediction of the effects of mutations on protein func- tion.Advances in neural information processing systems 34:29287–29303
Meier J, et al. (2021) Language models enable zero-shot prediction of the effects of mutations on protein func- tion.Advances in neural information processing systems 34:29287–29303
2021
-
[68]
(2022)Tranception: protein fitness predic- tion with autoregressive transformers and inference-time retrieval(PMLR), pp 16990–17017
Notin P, et al. (2022)Tranception: protein fitness predic- tion with autoregressive transformers and inference-time retrieval(PMLR), pp 16990–17017
2022
-
[69]
(2021) Optimization of therapeutic an- tibodies by predicting antigen specificity from antibody sequence via deep learning.Nature biomedical engineer- ing5:600–612
Mason DM, et al. (2021) Optimization of therapeutic an- tibodies by predicting antigen specificity from antibody sequence via deep learning.Nature biomedical engineer- ing5:600–612
2021
-
[70]
Pugh CW, Nu˜ nez-Valencia PG, Dias M, Frazer J (2026) From likelihood to fitness: Improving variant effect pre- diction in protein and genome language models.Advances in Neural Information Processing Systems38:130835– 130866
2026
-
[71]
(2026) Conditionally site-independent neu- ral evolution of antibody sequences.ArXivpp arXiv– 2602
Lu SZ, et al. (2026) Conditionally site-independent neu- ral evolution of antibody sequences.ArXivpp arXiv– 2602
2026
-
[72]
Molari M, Eyer K, Baudry J, Cocco S, Monasson R (2020) Quantitative modeling of the effect of antigen dosage on b-cell affinity distributions in maturating ger- minal centers.Elife9:e55678
2020
-
[73]
(2026) Inference of germinal center evo- lutionary dynamics via simulation-based deep learning
Ralph DK, et al. (2026) Inference of germinal center evo- lutionary dynamics via simulation-based deep learning. eLife14:RP108880
2026
-
[74]
Nucleic acids research41:W34–W40
Ye J, Ma N, Madden TL, Ostell JM (2013) Igblast: an immunoglobulin variable domain sequence analysis tool. Nucleic acids research41:W34–W40
2013
-
[75]
Gadala-Maria D, Yaari G, Uduman M, Kleinstein SH (2015) Automated analysis of high-throughput b-cell se- quencing data reveals a high frequency of novel im- munoglobulin v gene segment alleles.Proceedings of the National Academy of Sciences112:E862–E870
2015
-
[76]
Raybould MI, Kovaltsuk A, Marks C, Deane CM (2021) Cov-abdab: the coronavirus antibody database.Bioin- formatics37:734–735
2021
-
[77]
(2015) Mixcr: software for compre- hensive adaptive immunity profiling.Nature methods 12:380–381
Bolotin DA, et al. (2015) Mixcr: software for compre- hensive adaptive immunity profiling.Nature methods 12:380–381
2015
-
[78]
(2013) Models of somatic hypermutation targeting and substitution based on synonymous muta- tions from high-throughput immunoglobulin sequencing data.Frontiers in immunology4:358
Yaari G, et al. (2013) Models of somatic hypermutation targeting and substitution based on synonymous muta- tions from high-throughput immunoglobulin sequencing data.Frontiers in immunology4:358
2013
-
[79]
Spisak N, Walczak AM, Mora T (2020) Learning the heterogeneous hypermutation landscape of immunoglob- ulins from high-throughput repertoire data.Nucleic acids research48:10702–10712
2020
-
[80]
mikelov-et-al-2021
Good BH, Rouzine IM, Balick DJ, Hallatschek O, Desai MM (2012) Distribution of fixed beneficial mutations and the rate of adaptation in asexual populations.Proceed- ings of the National Academy of Sciences109:4950–4955. 17 Appendix A: Deep repertoire sequencing datasets The B cell repertoire datasets analyzed in this study fulfilled three criteria. Each s...
2012
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.