Molexar: A Unified Multimodal Molecular Foundation Model for Drug Design
Pith reviewed 2026-06-25 19:27 UTC · model grok-4.3
The pith
A single autoregressive decoder generates valid molecules from scalar properties, protein sequences, or binding pockets by replacing value-token embeddings during fine-tuning.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
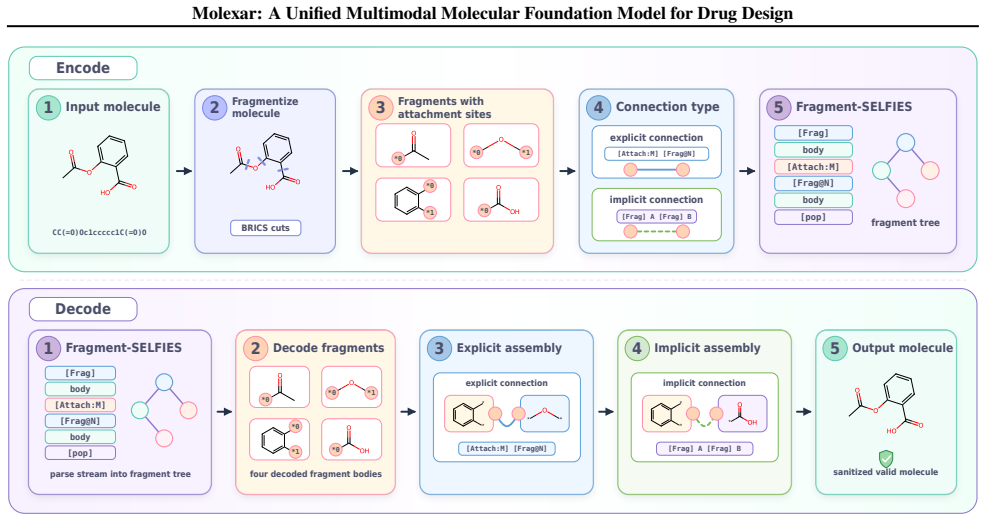
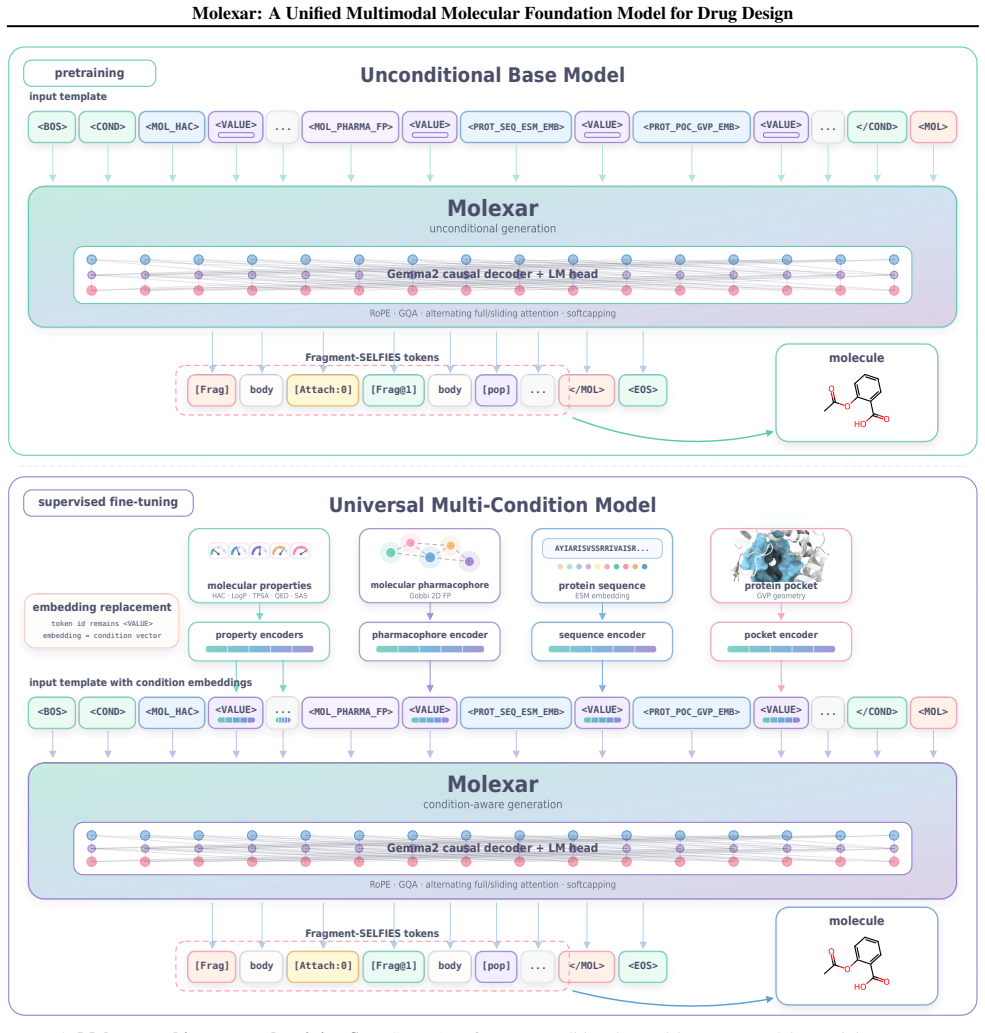
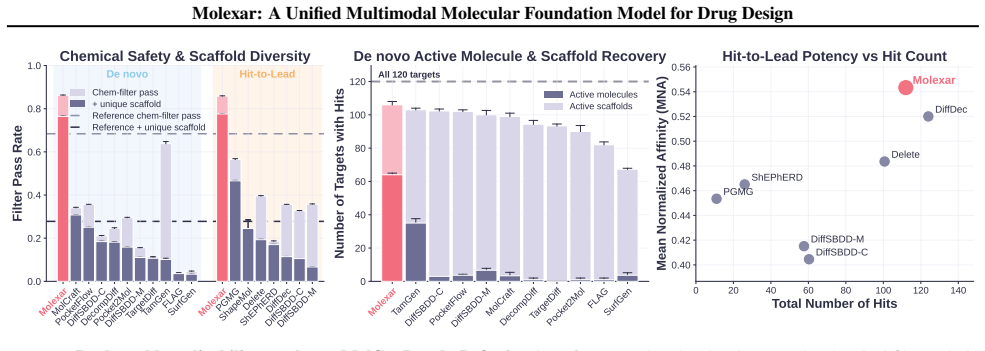
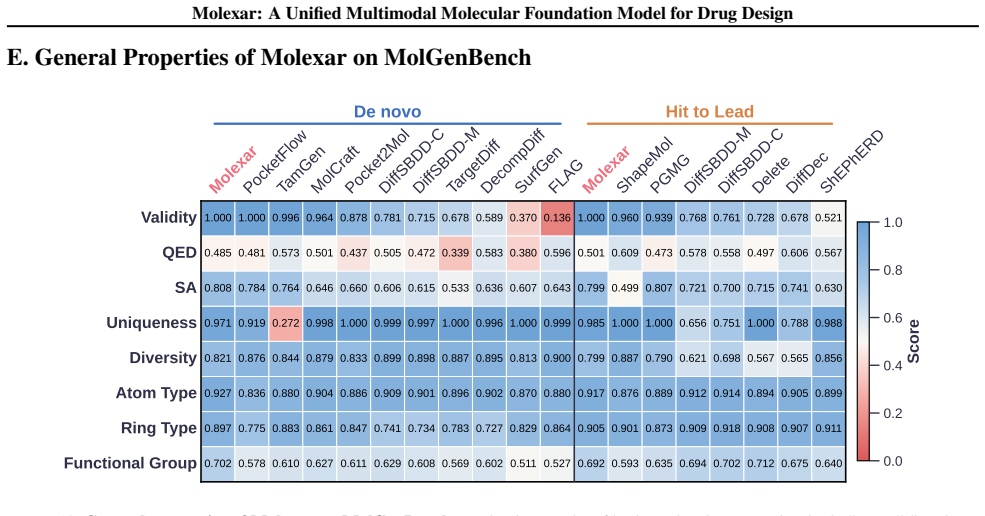
Molexar demonstrates that one small pretrained autoregressive decoder, trained on Fragment-SELFIES and then fine-tuned by in-place replacement of value-token embeddings for scalar properties, pharmacophore fingerprints, protein sequences, and binding pockets, reaches 100 percent validity, high drug-likeness, and competitive performance on CrossDocked2020 and MolGenBench tasks.
What carries the argument
In-place replacement of value-token embeddings during supervised fine-tuning, which lets one autoregressive decoder follow multiple conditioning modes without separate paths or architectures.
If this is right
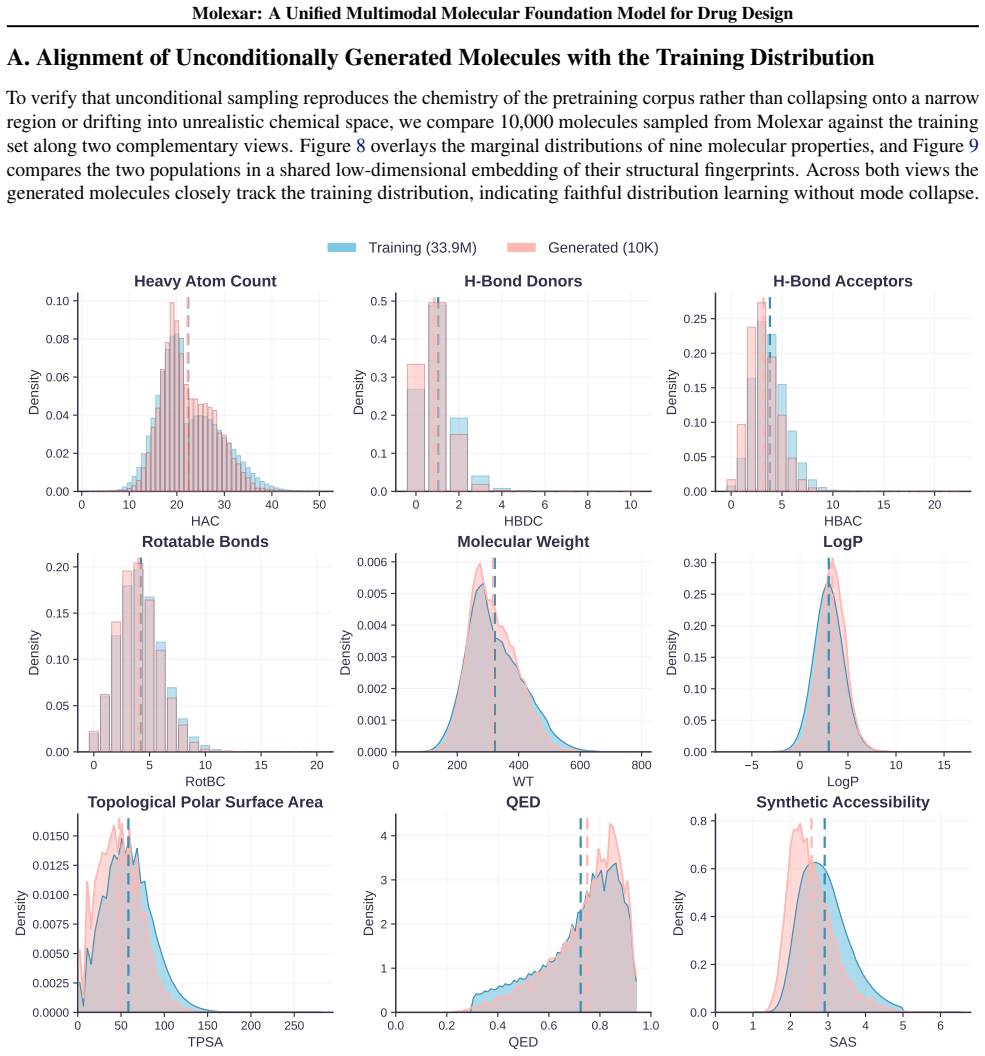
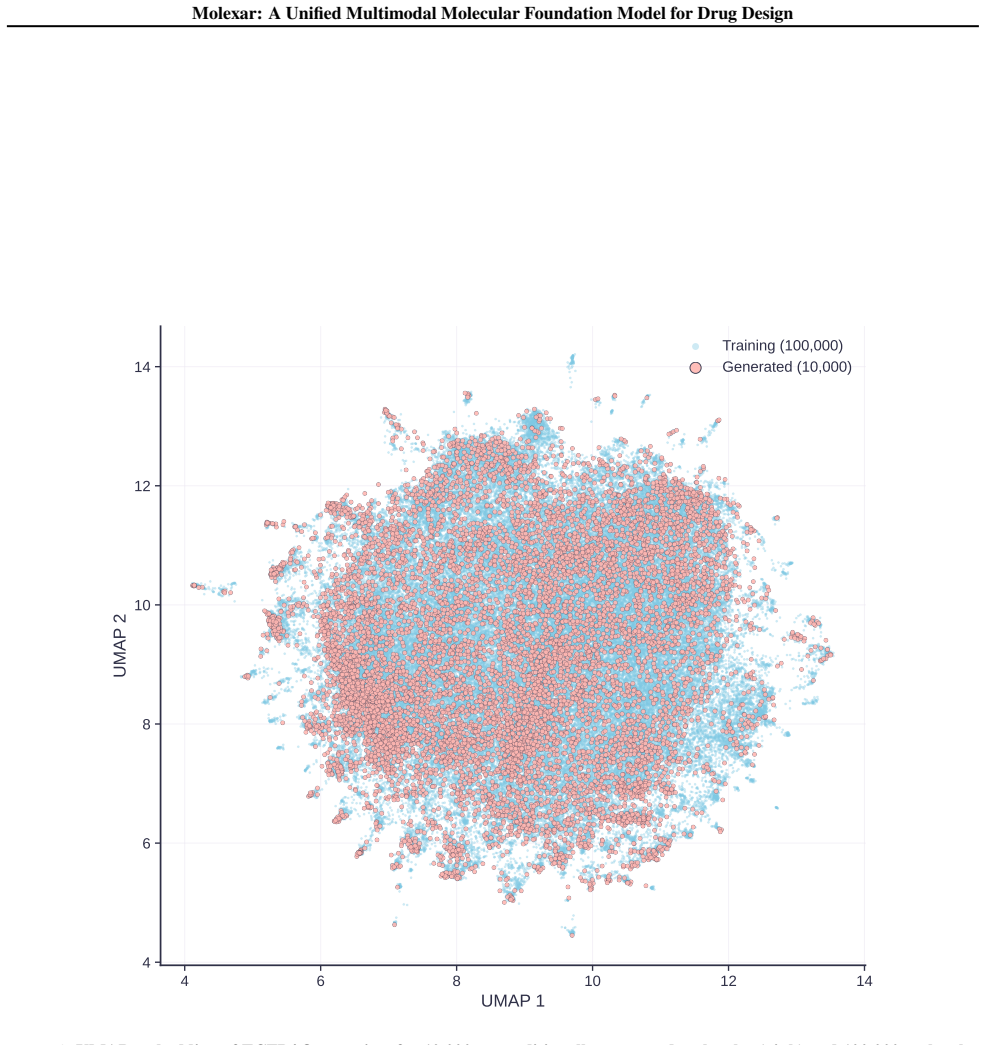
- The pretrained model alone already yields 100 percent validity and high drug-likeness for unconditional and fragment-constrained generation.
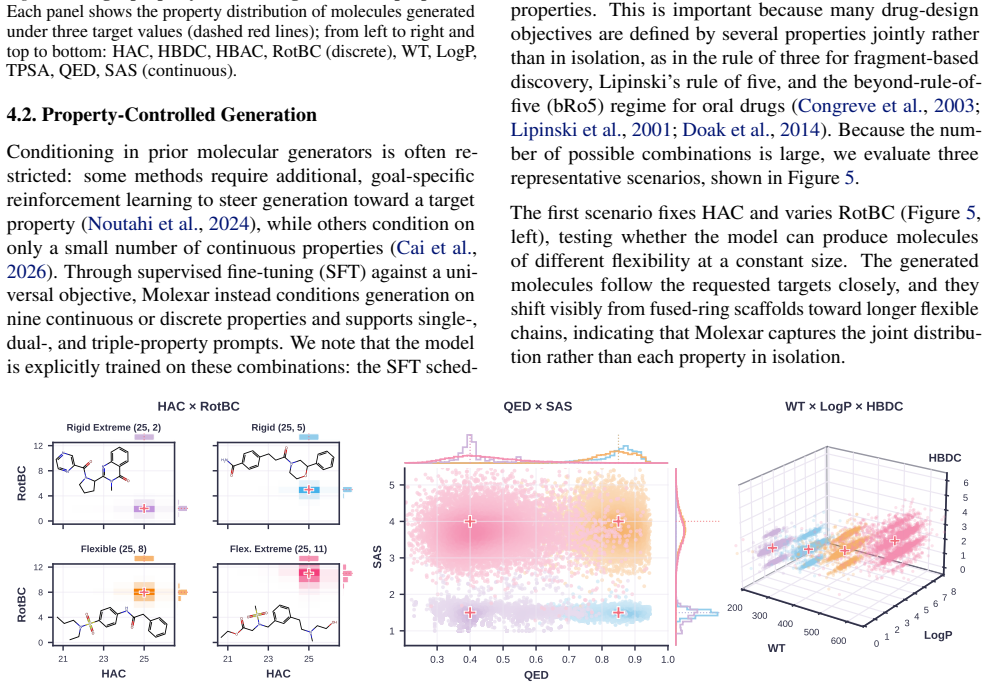
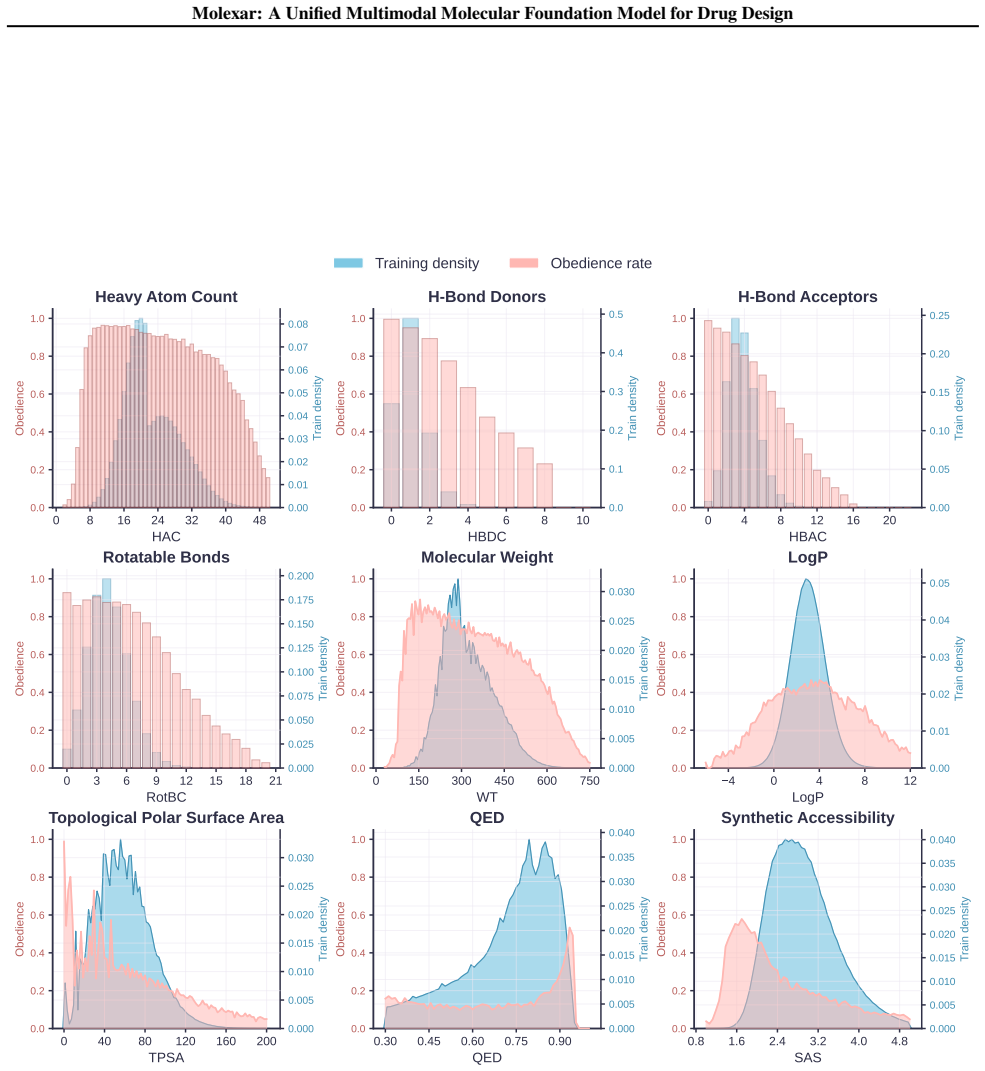
- The fine-tuned model follows both single-property and multi-property instructions while staying competitive on target-conditioned generation.
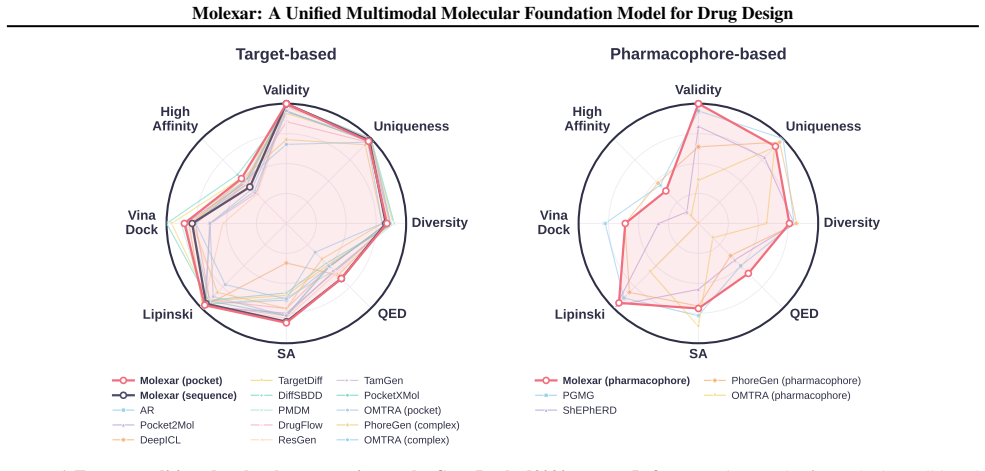
- Molecules produced on MolGenBench show favorable safety and potency profiles relative to other models.
- All modes share the same autoregressive generation path, eliminating the need for separate decoders per condition type.
Where Pith is reading between the lines
- The embedding-swap approach could be tested on additional condition types such as 3D conformer constraints without retraining the core decoder.
- Small-parameter models trained this way might be deployed directly in screening pipelines where larger multimodal systems are currently required.
- If the replacement trick scales cleanly, it reduces the engineering cost of adding new molecular design objectives to an existing foundation model.
Load-bearing premise
That swapping value-token embeddings in place is enough for the decoder to learn and follow every listed condition without losing validity or performance.
What would settle it
A test set where the model is given combined conditions such as a target protein sequence plus a required potency value and produces molecules that violate validity or fail to satisfy both constraints at rates higher than the reported baselines.
Figures



read the original abstract
Molecular generation is a central challenge in drug discovery, requiring models that explore vast chemical space while satisfying diverse design constraints. We present Molexar, a unified multimodal molecular foundation model built on Fragment-SELFIES, a robust, fragment-aware molecular language with validity-preserving decoding and explicit fragment structure. A pretrained autoregressive decoder learns the Fragment-SELFIES syntax and molecular distribution; supervised fine-tuning (SFT) then trains the same decoder on condition-molecule pairs spanning scalar molecular properties, pharmacophore fingerprints, protein sequences, and binding pockets, injecting each condition by in-place replacement of value-token embeddings so that all generation modes share one autoregressive path. Molexar achieves strong efficiency at a small parameter count while matching or exceeding larger models. The pretrained model reaches 100% validity and high drug-likeness in unconditional and fragment-constrained generation; the SFT model follows single- and multi-property instructions and remains competitive on target-conditioned generation on the CrossDocked2020 test set. On MolGenBench, Molexar further generates molecules with favorable safety and potency. These results establish Molexar as a practical unified foundation for computational chemistry and drug-design workflows.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper presents Molexar, a unified multimodal molecular foundation model using Fragment-SELFIES as input representation. A single autoregressive decoder is first pretrained on molecular strings for unconditional and fragment-constrained generation, then supervised fine-tuned on condition-molecule pairs (scalar properties, pharmacophore fingerprints, protein sequences, binding pockets) by injecting conditions via in-place replacement of value-token embeddings. The model is claimed to achieve 100% validity, high drug-likeness, competitive target-conditioned generation on CrossDocked2020, and favorable safety/potency on MolGenBench, all at small parameter count while matching or exceeding larger models.
Significance. If the central claims hold after detailed validation, the work would demonstrate a practical, parameter-efficient unified foundation model for multiple molecular generation modes in drug design. The Fragment-SELFIES representation and single-decoder conditioning strategy could simplify workflows; the reported 100% validity and competitive CrossDocked2020 results would be notable strengths if supported by reproducible code or data splits.
major comments (2)
- [Abstract / Methods] Abstract and methods description: the central conditioning mechanism (in-place replacement of value-token embeddings during SFT to handle scalar properties, pharmacophore fingerprints, protein sequences, and binding pockets within one autoregressive path) is described at high level only, with no equations, pseudocode, or ablation showing that this replacement preserves validity and performance across modes without interference or mode collapse. This is load-bearing for the unified multimodal claim.
- [Results] Results section: performance numbers (100% validity, competitive CrossDocked2020 scores, MolGenBench safety/potency) are stated without data splits, training hyperparameters, baseline details, error bars, or statistical tests, making it impossible to assess whether the small-parameter efficiency claim is supported or if results are reproducible.
minor comments (1)
- [Abstract] The abstract would benefit from explicit mention of model size (parameter count) and comparison models to ground the 'small parameter count' and 'matching or exceeding larger models' claims.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback. We address each major comment below and indicate the revisions that will be incorporated.
read point-by-point responses
-
Referee: [Abstract / Methods] Abstract and methods description: the central conditioning mechanism (in-place replacement of value-token embeddings during SFT to handle scalar properties, pharmacophore fingerprints, protein sequences, and binding pockets within one autoregressive path) is described at high level only, with no equations, pseudocode, or ablation showing that this replacement preserves validity and performance across modes without interference or mode collapse. This is load-bearing for the unified multimodal claim.
Authors: We agree that the current description of the in-place embedding replacement is high-level. In the revised manuscript we will expand the Methods section to include formal equations for the embedding replacement operation, pseudocode for the conditioned autoregressive generation procedure, and an ablation study that quantifies validity preservation and absence of mode collapse or cross-mode interference when conditioning on scalar properties, pharmacophores, proteins, and pockets. revision: yes
-
Referee: [Results] Results section: performance numbers (100% validity, competitive CrossDocked2020 scores, MolGenBench safety/potency) are stated without data splits, training hyperparameters, baseline details, error bars, or statistical tests, making it impossible to assess whether the small-parameter efficiency claim is supported or if results are reproducible.
Authors: We concur that the reported performance figures require additional supporting information for reproducibility and evaluation of the efficiency claims. The revised Results section will explicitly list the data splits employed for CrossDocked2020 and MolGenBench, all training hyperparameters, baseline configurations, error bars from repeated runs, and statistical significance tests. We will also release code and data splits to enable independent verification. revision: yes
Circularity Check
No significant circularity
full rationale
The paper describes an empirical architecture (Fragment-SELFIES language, pretrained autoregressive decoder, SFT via in-place value-token embedding replacement) and reports benchmark performance. No equations, fitted parameters renamed as predictions, self-citation load-bearing premises, uniqueness theorems, or ansatzes appear in the provided text. All claims reduce to standard training and evaluation procedures without internal reduction to the authors' prior definitions.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
URL https://openreview.net/forum? id=KSLkFYHlYg. Baell, J. B. and Holloway, G. A. New substructure fil- ters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclu- sion in bioassays.Journal of Medicinal Chemistry, 53 (7):2719–2740, 2010. doi: 10.1021/jm901137j. URL https://doi.org/10.1021/jm901137j. Bagal, V ...
-
[2]
URL https: //doi.org/10.1002/cmdc.200700139
doi: 10.1002/cmdc.200700139. URL https: //doi.org/10.1002/cmdc.200700139. Brown, N., Fiscato, M., Segler, M. H. S., and Vaucher, A. C. GuacaMol: Benchmarking models for de novo molecular design.Journal of Chemical Information and Modeling, 59(3):1096–1108, 2019. doi: 10.1021/acs.jcim. 8b00839. URL https://doi.org/10.1021/acs. jcim.8b00839. Bruns, R. F. an...
-
[3]
URL https: //doi.org/10.1039/D1SC02436A
doi: 10.1039/D1SC02436A. URL https: //doi.org/10.1039/D1SC02436A. Jadhav, A., Ferreira, R. S., Klumpp, C., Mott, B. T., Austin, C. P., Inglese, J., Thomas, C. J., Maloney, D. J., Shoichet, B. K., and Simeonov, A. Quantitative analyses of aggrega- tion, autofluorescence, and reactivity artifacts in a screen for inhibitors of a thiol protease.Journal of Med...
-
[4]
Jing, B., Eismann, S., Suriana, P., Townshend, R
URL https://proceedings.mlr.press/ v119/jin20b.html. Jing, B., Eismann, S., Suriana, P., Townshend, R. J. L., and Dror, R. O. Learning from protein structure with geometric vector perceptrons. InInternational Confer- ence on Learning Representations, 2021. URL https: //openreview.net/forum?id=1YLJDvSx6J4. Kallenborn, F., Chacon, A., Hundt, C., Sirelkhatim...
-
[5]
Lemos, P., Beckwith, Z., Bandi, S., van Damme, M., Crivelli-Decker, J., Shields, B
URL https://proceedings.mlr.press/ v267/lee25o.html. Lemos, P., Beckwith, Z., Bandi, S., van Damme, M., Crivelli-Decker, J., Shields, B. J., Merth, T., Jha, P. K., 14 Molexar: A Unified Multimodal Molecular Foundation Model for Drug Design De Mitri, N., Callahan, T. J., Nish, A., Abruzzo, P., Salomon-Ferrer, R., and Ganahl, M. SAIR: Enabling deep learning...
-
[6]
URL https://doi.org/10.1038/nprot. 2017.114. Li, Y ., Pei, J., and Lai, L. Structure-based de novo drug design using 3D deep generative models.Chemical Science, 12(41):13664–13675, 2021. doi: 10.1039/ D1SC04444C. URL https://doi.org/10.1039/ D1SC04444C. Lin, H., Huang, Y ., Zhang, O., Ma, S., Liu, M., Li, X., Wu, L., Wang, J., Hou, T., and Li, S. Z. DiffB...
-
[7]
Liu, Z., Li, Y ., Han, L., Li, J., Liu, J., Zhao, Z., Nie, W., Liu, Y ., and Wang, R
URL https://proceedings.mlr.press/ v162/liu22m.html. Liu, Z., Li, Y ., Han, L., Li, J., Liu, J., Zhao, Z., Nie, W., Liu, Y ., and Wang, R. PDB-wide collection of binding data: Current status of the PDBbind database. Bioinformatics, 31(3):405–412, 2015. doi: 10.1093/ bioinformatics/btu626. URL https://doi.org/10. 1093/bioinformatics/btu626. Luo, S., Guan, ...
2015
-
[8]
cc/paper_files/paper/2021/file/ 314450613369e0ee72d0da7f6fee773c-Paper
URL https://proceedings.neurips. cc/paper_files/paper/2021/file/ 314450613369e0ee72d0da7f6fee773c-Paper. pdf. Luo, Y ., Yang, K., Hong, M., Liu, X. Y ., and Nie, Z. MolFM: A multimodal molecular foundation model, 2023. URL https://arxiv.org/abs/2307.09484. arXiv preprint arXiv:2307.09484. Mysinger, M. M., Carchia, M., Irwin, J. J., and Shoichet, B. K. Dir...
-
[9]
Ragoza, M., Masuda, T., and Koes, D
URL https://proceedings.mlr.press/ v235/qu24a.html. Ragoza, M., Masuda, T., and Koes, D. R. Generating 3D molecules conditional on receptor binding sites with deep generative models.Chemical Science, 13(9):2701–2713,
-
[10]
URL https:// doi.org/10.1039/D1SC05976A
doi: 10.1039/D1SC05976A. URL https:// doi.org/10.1039/D1SC05976A. Schneuing, A., Harris, C., Du, Y ., Didi, K., Jamasb, A., Igashov, I., Du, W., Gomes, C., Blundell, T. L., Lio, P., Welling, M., Bronstein, M., and Correia, B. Structure- based drug design with equivariant diffusion models.Na- ture Computational Science, 4(12):899–909, 2024. doi: 10.1038/s4...
-
[11]
URL https://openreview.net/forum? id=g3VCIM94ke. Schuffenhauer, A., Schneider, N., Hintermann, S., Auld, D., Blank, J., Cotesta, S., Engeloch, C., Fechner, N., Gaul, C., Giovannoni, J., Jansen, J., Joslin, J., Krastel, P., Lounkine, E., Manchester, J., Monovich, L. G., Pellicci- oli, A. P., Schwarze, M., Shultz, M. D., Stiefl, N., and Baeschlin, D. K. Evo...
-
[12]
URL https: //doi.org/10.1038/s41467-025-56349-0
doi: 10.1038/s41467-025-56349-0. URL https: //doi.org/10.1038/s41467-025-56349-0. Zhang, O., Zhang, J., Jin, J., Zhang, X., Hu, R., Shen, C., Cao, H., Du, H., Kang, Y ., Deng, Y ., Liu, F., Chen, G., Hsieh, C.-Y ., and Hou, T. ResGen is a pocket-aware 3D molecular generation model based on parallel multiscale modelling.Nature Machine Intelligence, 5(9):1020–1030,
-
[13]
URL https: //doi.org/10.1038/s42256-023-00712-7
doi: 10.1038/s42256-023-00712-7. URL https: //doi.org/10.1038/s42256-023-00712-7. Zhu, H., Zhou, R., Cao, D., Tang, J., and Li, M. A pharmacophore-guided deep learning ap- proach for bioactive molecular generation.Nature Communications, 14(1):6234, 2023. doi: 10.1038/ s41467-023-41454-9. URL https://doi.org/10. 1038/s41467-023-41454-9. Zhung, W., Kim, H.,...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.