Commutative Algebra Learning for Protein Flexibility Analysis
Pith reviewed 2026-07-02 01:40 UTC · model grok-4.3
The pith
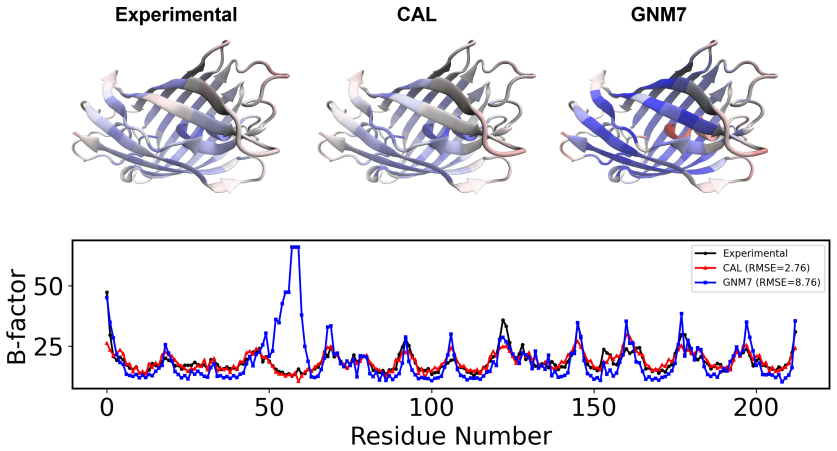
Commutative algebra constructs localized descriptors that raise B-factor prediction accuracy 34.5 percent above the Gaussian network model on 364 proteins.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
CAL employs commutative algebra theory to construct localized algebraic descriptors at multiple spatial scales; these descriptors accurately characterize the local geometric environments surrounding individual atoms and thereby improve B-factor prediction. On the benchmark of 364 proteins the approach yields a 34.5 percent accuracy increase relative to the Gaussian network model while remaining competitive with state-of-the-art methods; integration with machine learning further produces a blind cross-protein prediction model.
What carries the argument
localized algebraic descriptors at multiple spatial scales, built from commutative algebra theory to encode local atomic geometry
If this is right
- B-factor prediction improves on diverse protein datasets without requiring global structural features.
- The same localized descriptors support blind prediction across different proteins.
- The framework extends to other localized structural properties in biomolecular systems.
- Integration with existing machine-learning pipelines yields competitive performance while remaining mathematically interpretable.
Where Pith is reading between the lines
- The multi-scale algebraic construction may generalize to other atomic-resolution properties such as binding-site flexibility or allosteric pathways.
- If the descriptors remain stable under modest structural perturbations, they could serve as input features for dynamics simulations that start from static structures alone.
- Testing whether the same commutative-algebra pipeline improves predictions on nucleic acids or protein-nucleic acid complexes would reveal the breadth of the local-geometry encoding.
Load-bearing premise
Commutative algebra theory supplies localized descriptors at multiple scales that correctly capture the geometric environments around atoms and thereby determine B-factors.
What would settle it
A new set of proteins on which the algebraic descriptors produce no accuracy gain over the Gaussian network model, or on which the descriptors fail to distinguish local geometric differences that correlate with measured B-factors.
Figures






read the original abstract
Protein flexibility, commonly quantified by B-factors, is closely related to protein structure and function. However, accurate B-factor prediction remains challenging due to the multiscale nature of protein structures and the complexity of atomic interactions. In this work, we propose a commutative algebra-based learning framework, termed CAL, for protein B-factor prediction. Unlike many biomolecular prediction tasks that rely primarily on global structural representations, B-factor prediction requires an accurate characterization of the local geometric environments surrounding individual atoms. To address this challenge, CAL employs commutative algebra theory to construct localized algebraic descriptors at multiple spatial scales. On a benchmark dataset of 364 proteins, CAL improves prediction accuracy by 34.5\% over the classical Gaussian network model (GNM). Extensive experiments demonstrate that CAL achieves robust and consistent performance across diverse datasets and is competitive with existing state-of-the-art methods. Furthermore, by integrating CAL with machine learning, we develop a blind prediction model capable of cross-protein B-factor prediction. Overall, CAL provides an effective, efficient, and mathematically principled framework for protein flexibility prediction and offers a powerful approach for analyzing and predicting localized structural properties in complex biomolecular systems.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes CAL, a commutative algebra-based learning framework for predicting protein B-factors. It constructs localized algebraic descriptors at multiple spatial scales to characterize atomic geometric environments, reports a 34.5% accuracy improvement over the Gaussian network model (GNM) on a 364-protein benchmark, and integrates the descriptors with machine learning to enable blind cross-protein prediction.
Significance. If the central claims hold after full validation, the work would supply a mathematically principled alternative to physics-based network models for local flexibility analysis, with potential advantages in multiscale characterization and blind prediction. The reported quantitative gain and the emphasis on localized rather than global representations address a recognized gap in B-factor modeling.
major comments (3)
- [Abstract, §3] Abstract and §3 (Methods): the 34.5% improvement is stated without definition of the accuracy metric, the precise GNM baseline implementation, the train/test protocol on the 364-protein set, or any error bars or statistical tests; these omissions prevent verification that the data support the headline claim.
- [§2] §2 (Descriptor construction): the manuscript supplies no explicit construction of the commutative-algebra descriptors, including the choice of algebraic structures, the definition of the multiple spatial scales, or how localization around individual atoms is achieved; without these equations the weakest assumption cannot be evaluated.
- [§4] §4 (Machine-learning integration): the blind-prediction model is described only at a high level; the feature vector composition, training procedure, and cross-protein generalization protocol are not detailed, leaving open the possibility that performance gains arise from parameter fitting rather than the algebraic descriptors themselves.
minor comments (2)
- [§2] Notation for the algebraic descriptors is introduced without a clear table or appendix summarizing the symbols and their dimensions.
- [Abstract] The abstract states 'robust and consistent performance across diverse datasets' but does not list the additional datasets or report per-dataset statistics.
Simulated Author's Rebuttal
We thank the referee for their comments, which identify important omissions that affect the verifiability of our claims. We address each point below and will revise the manuscript to supply the requested details.
read point-by-point responses
-
Referee: [Abstract, §3] Abstract and §3 (Methods): the 34.5% improvement is stated without definition of the accuracy metric, the precise GNM baseline implementation, the train/test protocol on the 364-protein set, or any error bars or statistical tests; these omissions prevent verification that the data support the headline claim.
Authors: We agree that these elements were insufficiently specified. The 34.5% figure is the relative reduction in root-mean-square error versus GNM. GNM was implemented with the conventional 10 Å cutoff for the Kirchhoff matrix. The 364 proteins were partitioned 80/20 for training and testing, with results averaged over 5-fold cross-validation on the training portion. The revised manuscript will report standard deviations across 10 random seeds and p-values from a paired Wilcoxon test. revision: yes
-
Referee: [§2] §2 (Descriptor construction): the manuscript supplies no explicit construction of the commutative-algebra descriptors, including the choice of algebraic structures, the definition of the multiple spatial scales, or how localization around individual atoms is achieved; without these equations the weakest assumption cannot be evaluated.
Authors: We acknowledge that §2 presents the construction at a conceptual level without the full algebraic equations. The descriptors are obtained from the quotient ring of the polynomial ring generated by atomic coordinates inside concentric balls of radii 5 Å, 10 Å and 15 Å; localization is realized by restricting generators to the atom-centered filtration. The revised §2 will contain the explicit ring presentation, ideal generators, and feature-extraction map. revision: yes
-
Referee: [§4] §4 (Machine-learning integration): the blind-prediction model is described only at a high level; the feature vector composition, training procedure, and cross-protein generalization protocol are not detailed, leaving open the possibility that performance gains arise from parameter fitting rather than the algebraic descriptors themselves.
Authors: We will expand §4 to state that each atom’s feature vector is the concatenation of its three-scale algebraic descriptors, that a gradient-boosting regressor is trained by 5-fold cross-validation, and that blind evaluation uses leave-one-protein-out partitioning. Hyper-parameter selection was performed on an inner validation split; these additions will make clear that the reported gains derive from the descriptors rather than from model tuning alone. revision: yes
Circularity Check
No significant circularity identified
full rationale
The provided abstract describes a commutative-algebra framework for constructing localized descriptors at multiple scales, followed by integration with machine learning for B-factor prediction on a 364-protein benchmark, with a reported 34.5% accuracy gain over GNM. No equations, derivation steps, or self-citations are supplied that would allow inspection for self-definitional reductions, fitted inputs renamed as predictions, or load-bearing self-citation chains. The full manuscript text is referenced but not actually supplied in the query, preventing any concrete identification of a step that reduces to its own inputs by construction. The central claim therefore remains self-contained against external benchmarks and does not exhibit the enumerated circularity patterns.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
G.A. Petsko and D. Ringe.Protein Structure and Function. Primers in biology. New Science Press, 2004
work page 2004
- [2]
-
[3]
Hans Frauenfelder, Stephen G. Sligar, and Peter G. Wolynes. The energy landscapes and motions of proteins.Science, 254(5038):1598–1603, 1991
work page 1991
-
[4]
Nicole Hayes, Xiaoqi Wei, Hongsong Feng, Ekaterina Merkurjev, and Guo-Wei Wei. Persistent sheaf laplacian analysis of protein flexibility.The Journal of Physical Chemistry B, 129(17):4169–4178, April 2025
work page 2025
-
[5]
Trueblood, Hans-Beat B ¨urgi, Herbert Burzlaff, Jack D
Kenneth N. Trueblood, Hans-Beat B ¨urgi, Herbert Burzlaff, Jack D. Dunitz, Carlo M. Gramaccioli, Heinz H. Schulz, Uri Shmueli, and Sidney C. Abrahams. Atomic displacement parameter nomencla- ture report of a subcommittee on atomic displacement parameter nomenclature.Acta Crystallographica Section A: Foundations of Crystallography, 52(5):770–781, 1996
work page 1996
-
[6]
Zhoutong Sun, Qian Liu, Ge Qu, Yan Feng, and Manfred T. Reetz. Utility of b-factors in protein science: Interpreting rigidity, flexibility, and internal motion and engineering thermostability.Chemical Reviews, 119(3):1626–1665, January 2019
work page 2019
-
[7]
J. Andrew McCammon, Bruce R. Gelin, and Martin Karplus. Dynamics of folded proteins.Nature, 267(5612):585–590, 1977
work page 1977
-
[8]
Michael Levitt, Christian Sander, and Peter S. Stern. Protein normal-mode dynamics: Trypsin inhibitor, crambin, ribonuclease and lysozyme.Journal of Molecular Biology, 181(3):423–447, February 1985
work page 1985
-
[9]
Bernard R. Brooks, Robert E. Bruccoleri, Barry D. Olafson, David J. States, S. Swaminathan, and Martin Karplus. ¡scp¿charmm¡/scp¿: A program for macromolecular energy, minimization, and dynamics calculations.Journal of Computational Chemistry, 4(2):187–217, 1983
work page 1983
-
[10]
Coarse grained normal mode analysis vs
Jun-Koo Park, Robert Jernigan, and Zhijun Wu. Coarse grained normal mode analysis vs. refined gaus- sian network model for protein residue-level structural fluctuations.Bulletin of Mathematical Biology, 75(1):124–160, January 2013
work page 2013
-
[11]
Ivet Bahar, Ali Rana Atilgan, and Burak Erman. Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential.Folding and Design, 2(3):173–181, 1997
work page 1997
-
[12]
Ivet Bahar, Ali Rana Atilgan, Melik C. Demirel, and Burak Erman. Vibrational dynamics of folded proteins: Significance of slow and fast motions in relation to function and stability.Physical Review Letters, 80(12):2733–2736, March 1998
work page 1998
-
[13]
A.R. Atilgan, S.R. Durell, R.L. Jernigan, M.C. Demirel, O. Keskin, and I. Bahar. Anisotropy of fluctu- ation dynamics of proteins with an elastic network model.Biophysical Journal, 80(1):505–515, January 2001
work page 2001
-
[14]
Kristopher Opron, Kelin Xia, and Guo-Wei Wei. Communication: Capturing protein multiscale ther- mal fluctuations.The Journal of Chemical Physics, 142(21), 2015. 18
work page 2015
-
[15]
Kelin Xia, Kristopher Opron, and Guo-Wei Wei. Multiscale multiphysics and multidomain mod- els—flexibility and rigidity.The Journal of Chemical Physics, 139(19), November 2013
work page 2013
-
[16]
Stochastic model for protein flexibility analysis.Physical Review E, 88(6), December 2013
Kelin Xia and Guo-Wei Wei. Stochastic model for protein flexibility analysis.Physical Review E, 88(6), December 2013
work page 2013
-
[17]
Kristopher Opron, Kelin Xia, and Guo-Wei Wei. Fast and anisotropic flexibility-rigidity index for protein flexibility and fluctuation analysis.The Journal of Chemical Physics, 140(23), 2014
work page 2014
-
[18]
Hongsong Feng, Jeffrey Y. Zhao, and Guo-Wei Wei. Multiscale differential geometry learning for pro- tein flexibility analysis.Journal of Computational Chemistry, 46(7), March 2025
work page 2025
-
[19]
Wanying Bi, Hongsong Feng, Jie Wu, Jingyan Li, and Guo-Wei Wei. Topological magnitude for protein flexibility analysis.Royal Society Open Science, 12(12), December 2025
work page 2025
-
[20]
Persistent stanley–reisner theory.Foundations of Data Science, 8:287–312, 2026
Faisal Suwayyid and Guo-Wei Wei. Persistent stanley–reisner theory.Foundations of Data Science, 8:287–312, 2026
work page 2026
-
[21]
Rui Wang, Duc Duy Nguyen, and Guo-Wei Wei. Persistent spectral graph.International Journal for Numerical Methods in Biomedical Engineering, 36(9):e3376, 2020
work page 2020
-
[22]
Li Shen, Jian Liu, and Guo-Wei Wei. Persistent mayer homology and persistent mayer laplacian.Foun- dations of Data Science, 6(4):584–612, 2024
work page 2024
-
[23]
Local laplacian: theory and models for data analysis.arXiv preprint arXiv:2603.07591, 2026
Jian Liu, Hongsong Feng, and Kefeng Liu. Local laplacian: theory and models for data analysis.arXiv preprint arXiv:2603.07591, 2026
-
[24]
Facundo M ´emoli, Zhengchao Wan, and Yusu Wang. Persistent laplacians: Properties, algorithms and implications.SIAM Journal on Mathematics of Data Science, 4(2):858–884, 2022
work page 2022
-
[25]
Hongsong Feng and Guo-Wei Wei. Virtual screening of drugbank database for herg blockers using topological laplacian-assisted ai models.Computers in biology and medicine, 153:106491, 2023
work page 2023
-
[26]
Persistent local laplacian prediction of protein-ligand binding affinities
Jian Liu and Hongsong Feng. Persistent local laplacian prediction of protein-ligand binding affinities. arXiv preprint arXiv:2603.21503, 2026
-
[27]
Hongsong Feng, Faisal Suwayyid, Mushal Zia, JunJie Wee, Yuta Hozumi, Chun-Long Chen, and Guo- Wei Wei. Caml: Commutative algebra machine learning-a case study on protein–ligand binding affin- ity prediction.Journal of Chemical Information and Modeling, 65(13):6732–6743, 2025
work page 2025
-
[28]
Mushal Zia, Faisal Suwayyid, Yuta Hozumi, JunJie Wee, Hongsong Feng, and Guo-Wei Wei. Cap: Commutative algebra prediction of protein-nucleic acid binding affinities.Machine Learning: Science and T echnology, 6(4):045068, 2025
work page 2025
-
[29]
JunJie Wee, Faisal Suwayyid, Mushal Zia, Hongsong Feng, Yuta Hozumi, and Guo-Wei Wei. Com- mutative algebra neural network reveals genetic origins of diseases.arXiv preprint arXiv:2509.26566, 2025
-
[30]
Cakl: Commutative algebra k-mer learning of genomics,
Faisal Suwayyid, Yuta Hozumi, Hongsong Feng, Mushal Zia, JunJie Wee, and Guo-Wei Wei. Cakl: Commutative algebra k-mer learning of genomics.arXiv preprint arXiv:2508.09406, 2025
-
[31]
Caleb Simiyu Khaemba, Hongsong Feng, Dong Chen, Chun-Long Chen, and Guo-Wei Wei. Commu- tative algebra modeling in materials science–a case study on metal–organic frameworks (mofs).Journal of Chemical Information and Modeling, 66(5):2584–2596, 2026
work page 2026
-
[32]
David Bramer and Guo-Wei Wei. Atom-specific persistent homology and its application to protein flexibility analysis.Computational and Mathematical Biophysics, 8(1):1–35, January 2020. 19
work page 2020
-
[33]
M. Heinig and D. Frishman. Stride: a web server for secondary structure assignment from known atomic coordinates of proteins.Nucleic Acids Research, 32(Web Server):W500–W502, 2004
work page 2004
-
[34]
Vmd: Visual molecular dynamics.Journal of Molecular Graphics, 14(1):33–38, February 1996
William Humphrey, Andrew Dalke, and Klaus Schulten. Vmd: Visual molecular dynamics.Journal of Molecular Graphics, 14(1):33–38, February 1996. 20
work page 1996
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.