Shared quasispecies architecture in experimental and natural RNA virus populations
Pith reviewed 2026-06-30 21:20 UTC · model grok-4.3
The pith
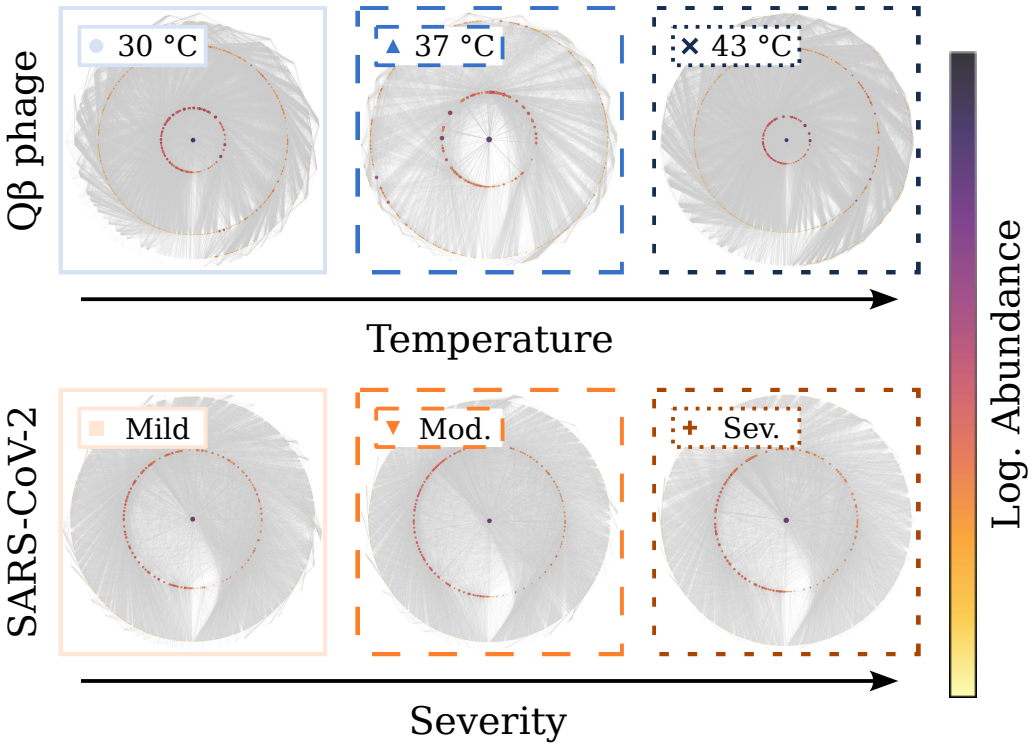
RNA viruses from lab phage and human coronavirus share a genotype network with one dominant central haplotype ringed by layers of rarer variants.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
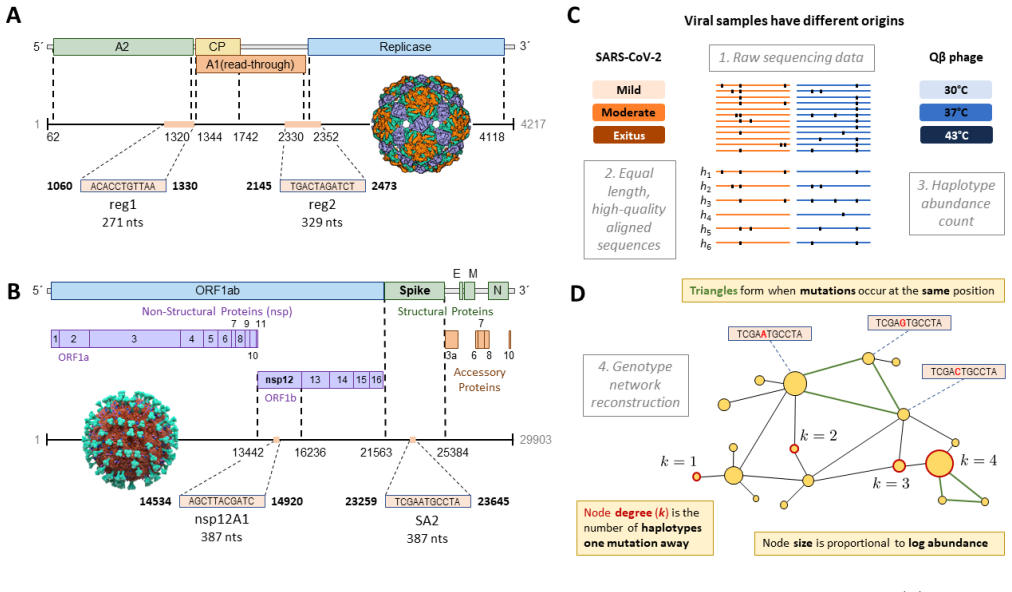
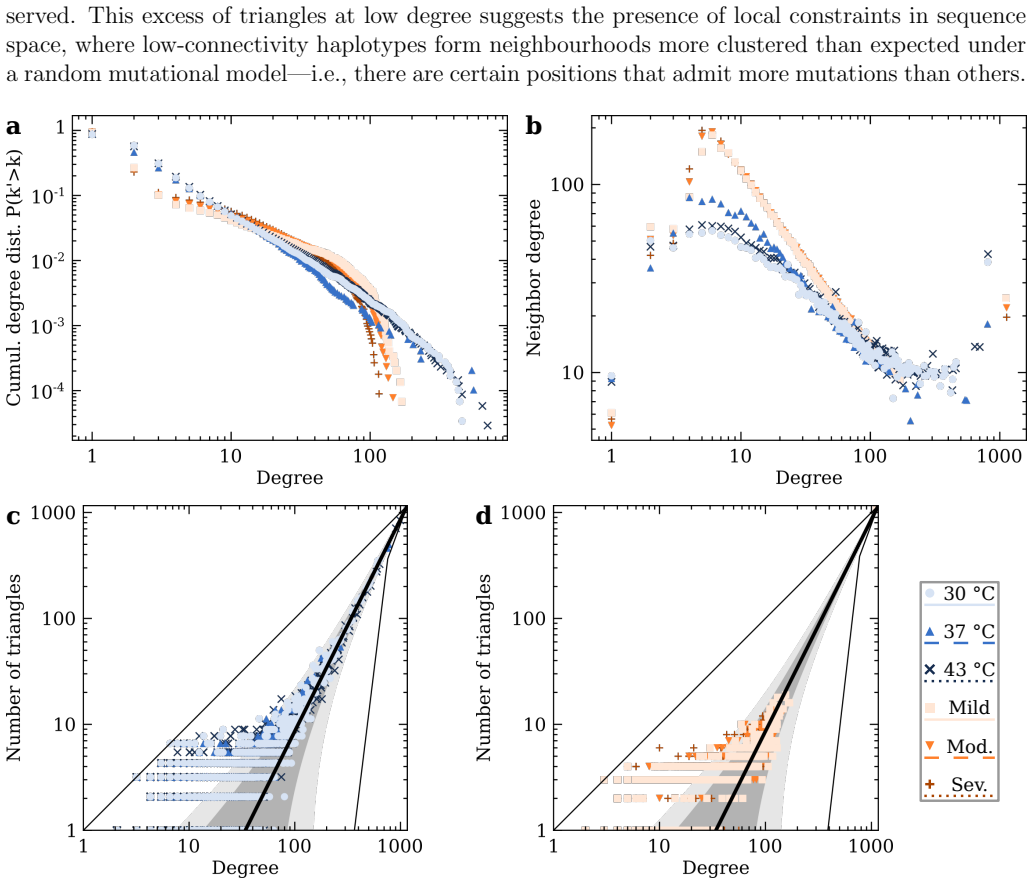
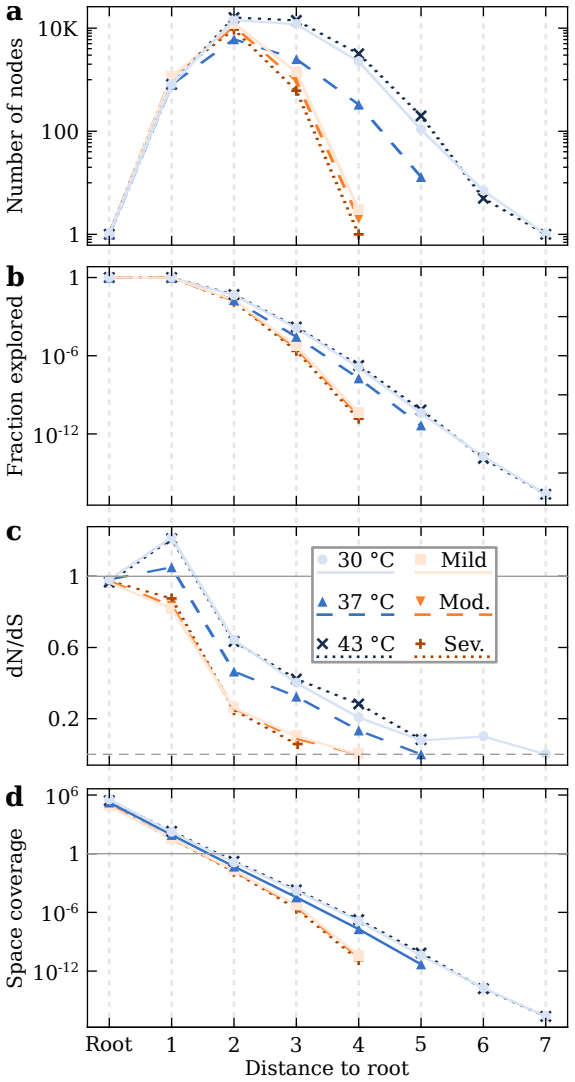
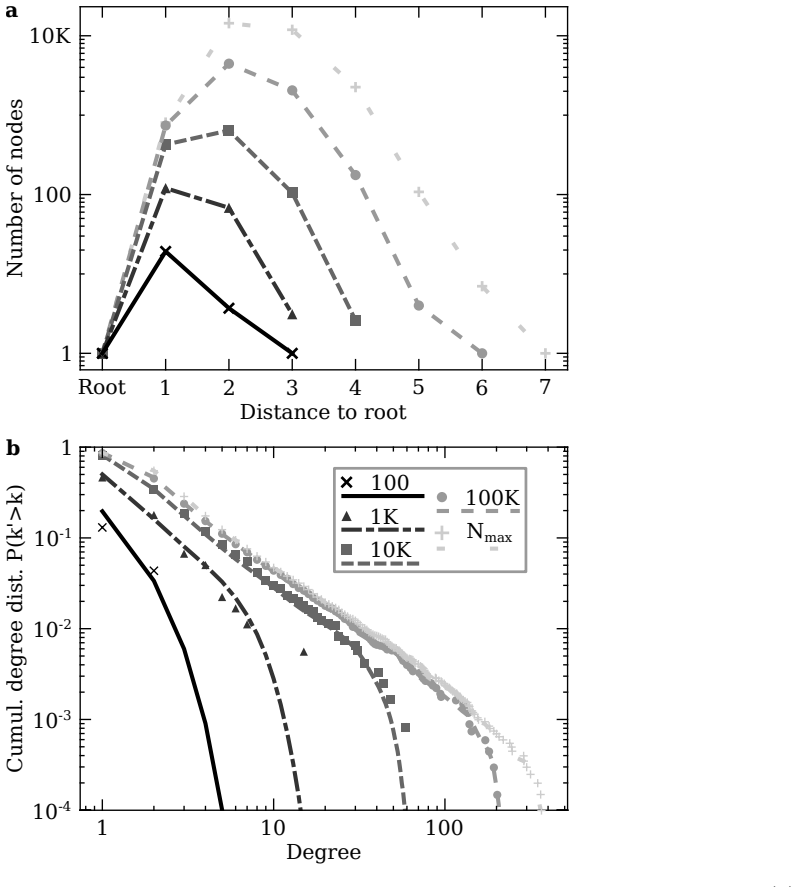
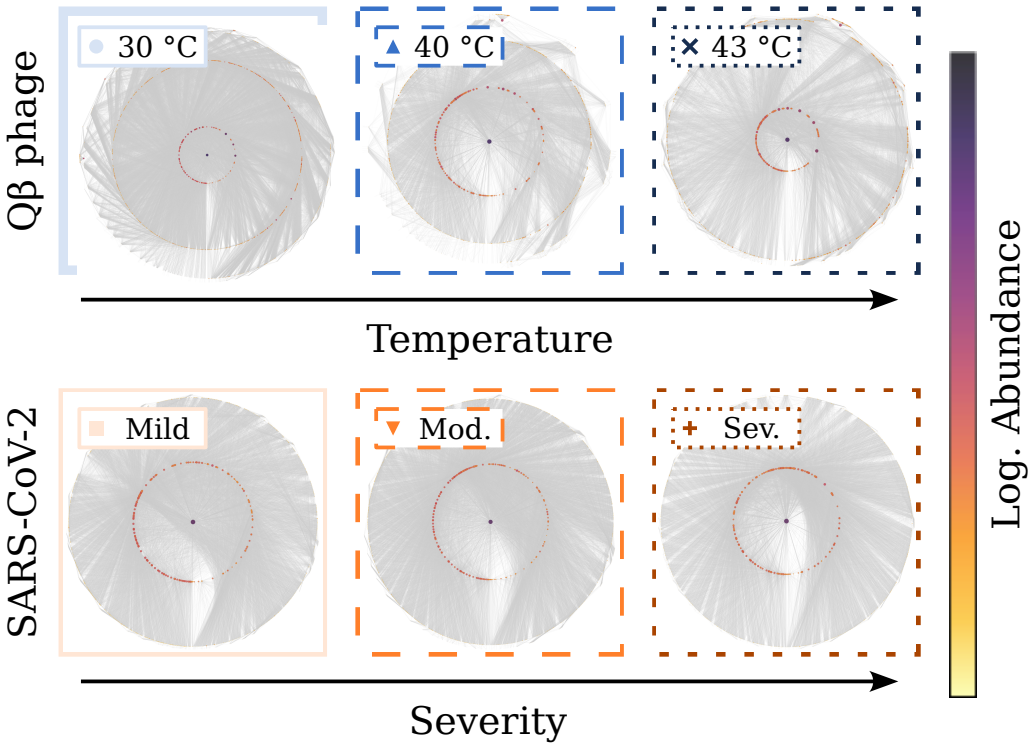
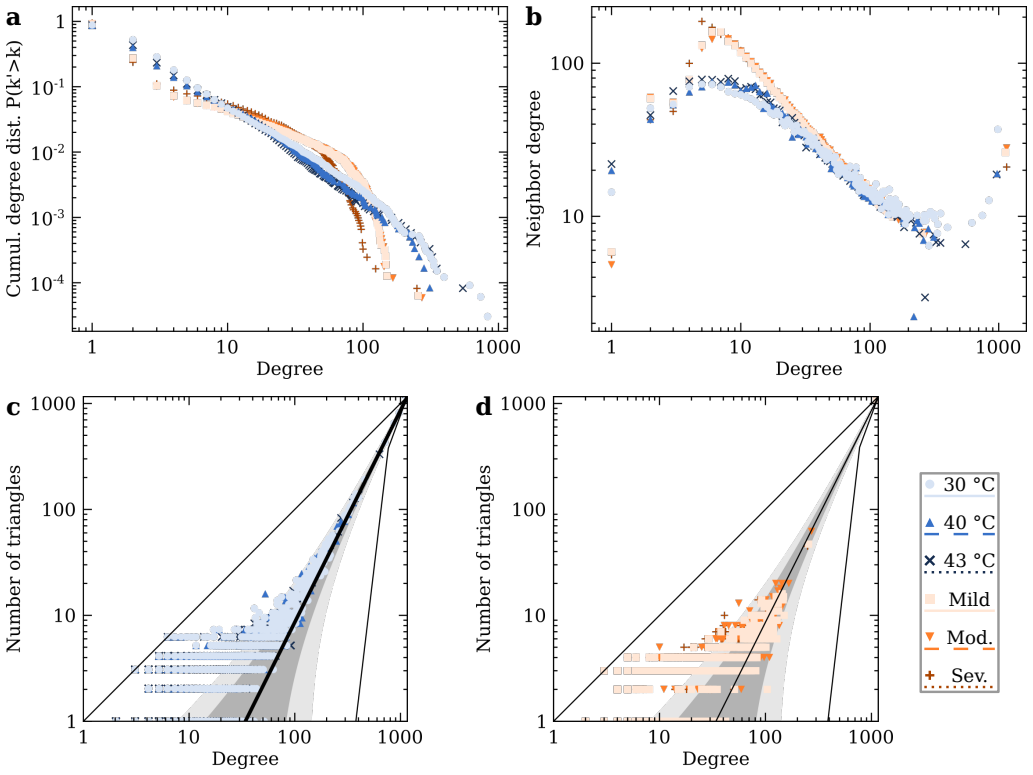
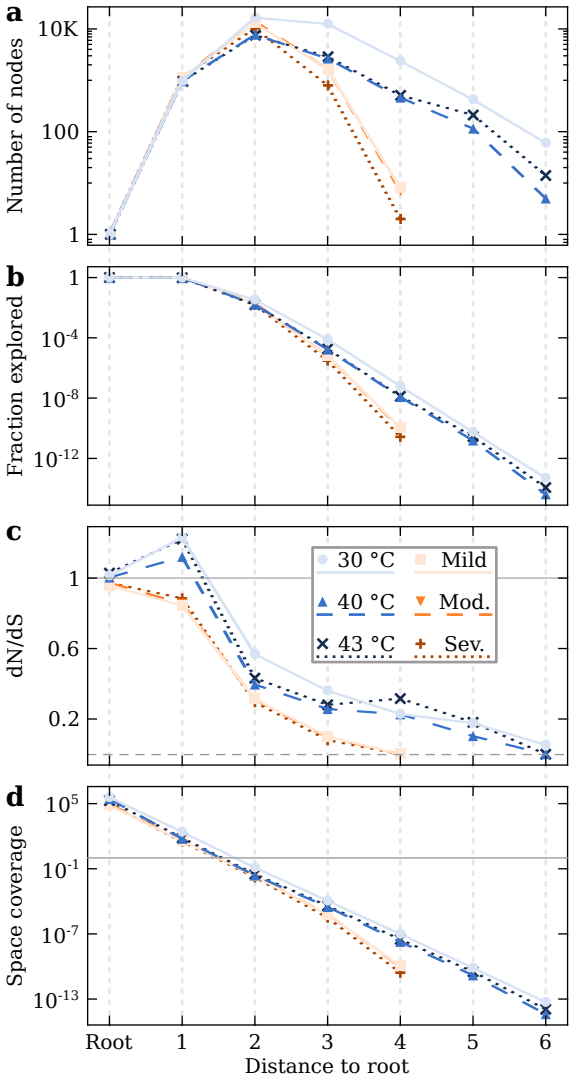
RNA viruses form genetically diverse populations structured as mutant spectra whose internal organization is captured by genotype networks. Deep sequencing of Qβ bacteriophage and SARS-CoV-2 populations reveals that both exhibit a highly abundant central haplotype surrounded by layers of variants of diminishing abundance as Hamming distance to the central haplotype increases. All reconstructed networks share qualitative and quantitative topological features and display a hierarchical structure. The robust organization under multiple conditions indicates that RNA viruses share a common genotype network architecture governed by fundamental properties of sequence space and the generic mechanism
What carries the argument
Genotype network: the graph of mutationally connected variants whose nodes are labeled by observed abundance, which organizes the population into a central high-abundance haplotype with concentric shells of lower-abundance mutants.
If this is right
- Genotype networks supply a unifying description of viral population structure that goes beyond standard diversity statistics.
- Local constraints in sequence space shape the mutational search available to the population and thereby affect evolutionary predictability.
- The same hierarchical pattern should appear in other RNA viruses whenever replication and mutation act on comparable sequence spaces.
- The architecture remains stable across laboratory and natural host settings, implying that basic replication rules dominate over ecological differences.
Where Pith is reading between the lines
- If the layered structure is general, targeted sequencing of early-pandemic or vaccine-breakthrough samples could test whether the central haplotype shifts in predictable ways.
- The pattern may extend to other high-mutation replicating systems such as certain RNA bacteriophages or segmented viruses, offering a comparative test.
- Network topology could be used to forecast which low-frequency variants are most likely to reach high abundance under new selective pressures.
Load-bearing premise
Deep-sequencing reads accurately reconstruct the true genotype network without distortion from PCR bias, sequencing errors, or incomplete sampling of rare variants.
What would settle it
Reconstructing genotype networks from deep sequencing of additional RNA virus populations and finding that variant abundance does not decrease with Hamming distance from a central haplotype would falsify the shared-architecture claim.
Figures
read the original abstract
RNA viruses form genetically diverse populations structured as mutant spectra, or quasispecies, whose internal organization influences their evolutionary and adaptive dynamics. While genetic diversity has been extensively characterized, the structural organization of viral populations in sequence space remains less explored. Here, we compare genotype network architectures in two RNA viruses with markedly different evolutionary contexts: bacteriophage $Q\beta$ evolving in controlled laboratory conditions and SARS-CoV-2 evolving within infected human hosts. Using deep sequencing data, we reconstruct the genotype network of mutationally coupled variants within viral populations and analyze their topological properties. Despite large differences in genome size, mutation rate, and ecological setting, both viruses exhibit a common organization: a highly abundant central haplotype surrounded by layers of variants of diminishing abundance as Hamming distance to the central haplotype increases. All reconstructed networks share qualitative and quantitative topological features, displaying a hierarchical structure. The robust organization of both populations under multiple conditions suggests that RNA viruses may share a common genotype network architecture governed by fundamental properties of sequence space and the generic mechanisms of replication and mutation. Genotype networks provide a unifying framework to describe viral population structure beyond conventional diversity measures and, by revealing how local constraints shape mutational search, offers insights into the predictability of viral evolution.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript analyzes deep-sequencing data from bacteriophage Qβ (laboratory evolution) and SARS-CoV-2 (clinical samples) to reconstruct genotype networks of mutationally coupled variants. It claims that, despite large differences in genome size, mutation rate, and ecological setting, both viruses display a shared quasispecies architecture: a highly abundant central haplotype surrounded by successive layers of lower-abundance variants ordered by increasing Hamming distance, with all networks sharing qualitative and quantitative topological features indicative of a hierarchical structure. The authors conclude that this organization is governed by fundamental properties of sequence space and generic replication/mutation mechanisms, providing a unifying framework beyond conventional diversity measures.
Significance. If the reported central-haplotype-plus-diminishing-abundance pattern is shown to be robust to sequencing artifacts, the result would supply a concrete, falsifiable description of intra-host population structure that could improve models of mutational exploration and evolutionary predictability across RNA viruses. The cross-system comparison is potentially valuable because the two viruses differ substantially in scale and context; however, the absence of methodological controls prevents any assessment of whether the claimed commonality reflects biology or data-generation biases.
major comments (2)
- [Abstract] Abstract: the description of network reconstruction supplies no information on read-depth thresholds, error-correction procedures, network-construction algorithm, statistical tests against null models, or controls for sequencing artifacts, rendering it impossible to judge whether the data support the central claim.
- [Abstract] The load-bearing assumption that the reconstructed networks faithfully represent true intra-host populations is not addressed. Deep sequencing of RNA viruses is known to be distorted by PCR jackpotting (which inflates certain haplotypes), base-calling errors (which preferentially populate low-frequency bins at Hamming distance 1–3), and uneven coverage (which undersamples rare variants). Because the reported architecture is defined quantitatively by abundance ordered by Hamming distance, any systematic bias correlated with these quantities will generate the observed pattern even if the underlying biology differs.
Simulated Author's Rebuttal
We thank the referee for their constructive comments highlighting the need for greater methodological transparency in the abstract and for raising important questions about potential sequencing artifacts. We address each point below and have revised the manuscript to improve clarity and strengthen the presentation of controls.
read point-by-point responses
-
Referee: [Abstract] Abstract: the description of network reconstruction supplies no information on read-depth thresholds, error-correction procedures, network-construction algorithm, statistical tests against null models, or controls for sequencing artifacts, rendering it impossible to judge whether the data support the central claim.
Authors: We agree that the abstract, owing to length constraints, did not summarize these parameters. The full Methods section already specifies read-depth thresholds (minimum 500–1000 reads per haplotype), error correction via Phred-score filtering and consensus calling, network construction by connecting haplotypes at Hamming distance 1, and comparisons to null models generated by randomizing variant abundances while preserving total diversity. To address the referee’s concern directly, we have expanded the abstract to include concise statements of these elements and the artifact controls (down-sampling and simulated error profiles). revision: yes
-
Referee: [Abstract] The load-bearing assumption that the reconstructed networks faithfully represent true intra-host populations is not addressed. Deep sequencing of RNA viruses is known to be distorted by PCR jackpotting (which inflates certain haplotypes), base-calling errors (which preferentially populate low-frequency bins at Hamming distance 1–3), and uneven coverage (which undersamples rare variants). Because the reported architecture is defined quantitatively by abundance ordered by Hamming distance, any systematic bias correlated with these quantities will generate the observed pattern even if the underlying biology differs.
Authors: This concern is substantive. While the original abstract did not explicitly discuss bias mitigation, the manuscript already contains (i) high-coverage Qβ laboratory data generated with multiple independent RT-PCR replicates to reduce jackpotting, (ii) application of the same error-correction pipeline to both Qβ and SARS-CoV-2 datasets, and (iii) statistical tests showing that the observed abundance–distance relationship deviates significantly from expectations under uniform or PCR-biased null models. In revision we have added an explicit paragraph in the Discussion that quantifies the expected impact of base-calling errors and uneven coverage and demonstrates that these artifacts alone cannot reproduce the hierarchical layering seen in both systems. We therefore maintain that the shared architecture is unlikely to be an artifact, but we acknowledge that orthogonal validation (e.g., single-molecule sequencing) would further strengthen the claim. revision: partial
Circularity Check
No circularity: empirical data observation
full rationale
The paper reports direct observations from deep sequencing of Qβ and SARS-CoV-2 populations, reconstructing genotype networks and noting a shared hierarchical pattern of central high-abundance haplotypes with diminishing-abundance shells by Hamming distance. No equations, fitted parameters, or derivations are presented that reduce to inputs by construction. The central claim is an empirical description of measured topological features, not a prediction or uniqueness result derived from self-citation or ansatz. The analysis stands as a self-contained comparison of two independent datasets.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Deep sequencing data can be used to reconstruct the true genotype network of a viral population without material distortion from amplification bias, sequencing errors, or sampling incompleteness.
Reference graph
Works this paper leans on
-
[1]
The architecture of an empirical genotype-phenotype map
Jos´ e Aguilar-Rodr´ ıguez et al. “The architecture of an empirical genotype-phenotype map”. In: Evolution72.6 (2018), pp. 1242–1260
2018
-
[2]
Topological structure of the space of phenotypes: the case of RNA neutral networks
Jacobo Aguirre et al. “Topological structure of the space of phenotypes: the case of RNA neutral networks”. In:PLoS ONE6 (2011), e26324
2011
-
[3]
Raul Andino and Esteban Domingo. “Viral quasispecies”. In:Virology479-480 (2015). 60th An- niversary Issue, pp. 46–51.issn: 0042-6822.doi:https://doi.org/10.1016/j.virol.2015. 03.022.url:https://www.sciencedirect.com/science/article/pii/S0042682215001580
-
[4]
Intra-Population Competition during Adaptation to In- creased Temperature in an RNA Bacteriophage
Mar´ ıa Arribas and Ester L´ azaro. “Intra-Population Competition during Adaptation to In- creased Temperature in an RNA Bacteriophage”. In:International Journal of Molecular Sci- ences22 (June 2021), p. 6815.doi:10.3390/ijms22136815
-
[5]
Adaptation to Fluctuating Temperatures in an RNA Virus Is Driven by the Most Stringent Selective Pressure
Mar´ ıa Arribas et al. “Adaptation to Fluctuating Temperatures in an RNA Virus Is Driven by the Most Stringent Selective Pressure”. In:PloS one9 (June 2014), e100940.doi:10.1371/ journal.pone.0100940. 16
2014
-
[6]
SARS-CoV-2 biological clones are genetically heterogeneous and include clade-discordant residues
Ana Isabel de ´Avila et al. “SARS-CoV-2 biological clones are genetically heterogeneous and include clade-discordant residues”. In:Journal of Virology99.5 (2025), e02250–24.doi:10. 1128/jvi.02250-24. eprint:https://journals.asm.org/doi/pdf/10.1128/jvi.02250-24. url:https://journals.asm.org/doi/abs/10.1128/jvi.02250-24
-
[7]
The proportion of revertant and mutant phage in a growing population, as a function of mutation and growth rate
Eduard Batschelet, Esteban Domingo, and Charles Weissmann. “The proportion of revertant and mutant phage in a growing population, as a function of mutation and growth rate”. In: Gene1.1 (1976), pp. 27–32.issn: 0378-1119.doi:https : / / doi . org / 10 . 1016 / 0378 - 1119(76 ) 90004 - 4.url:https : / / www . sciencedirect . com / science / article / pii / ...
1976
-
[8]
RNA virus population diver- sity: implications for inter-species transmission
Antonio V Border´ ıa, Kenneth A Stapleford, and Marco Vignuzzi. “RNA virus population diver- sity: implications for inter-species transmission”. In:Current Opinion in Virology1.6 (2011). In- nate immunity/Antivirals and resistance/Emerging viruses, pp. 643–648.issn: 1879-6257.doi: https://doi.org/10.1016/j.coviro.2011.09.012.url:https://www.sciencedirect....
work page doi:10.1016/j.coviro.2011.09.012.url:https://www.sciencedirect 2011
-
[9]
Incipient functional SARS-CoV-2 diversification identified through neural network haplotype maps
Soledad Delgado et al. “Incipient functional SARS-CoV-2 diversification identified through neural network haplotype maps”. In:Proceedings of the National Academy of Sciences121.10 (2024), e2317851121.doi:10.1073/pnas.2317851121. eprint:https://www.pnas.org/doi/ pdf/10.1073/pnas.2317851121.url:https://www.pnas.org/doi/abs/10.1073/pnas. 2317851121
-
[10]
Viral Quasispecies Evolution
Esteban Domingo, Julie Sheldon, and Celia Perales. “Viral Quasispecies Evolution”. In:Mi- crobiology and Molecular Biology Reviews76.2 (2012), pp. 159–216
2012
-
[11]
Nucleotide sequence heterogeneity of an RNA phage population
Esteban Domingo et al. “Nucleotide sequence heterogeneity of an RNA phage population”. In:Cell13.4 (1978), pp. 735–744.issn: 0092-8674.doi:https://doi.org/10.1016/0092- 8674(78)90223-4
-
[12]
Selforganization of matter and the evolution of biological macromolecules
Manfred Eigen. “Selforganization of matter and the evolution of biological macromolecules”. In:Naturwissenschaften58 (1971), pp. 465–523
1971
-
[13]
Analysis of RNA sequence structure maps by exhaustive enumeration II. Structures of neutral networks and shape space covering
Walter Gr¨ uner et al. “Analysis of RNA sequence structure maps by exhaustive enumeration II. Structures of neutral networks and shape space covering”. In:Monatsh. Chem.127 (1996), pp. 375–389
1996
-
[14]
Hugh K Haddox et al. “Clonal interference and changing selective pressures shape the escape of SARS-CoV-2 from hundreds of antibodies”. In:Virus Evolution12.1 (Feb. 2026), veaf104.issn: 2057-1577.doi:10.1093/ve/veaf104. eprint:https://academic.oup.com/ve/article- pdf/12/1/veaf104/66747267/veaf104.pdf.url:https://doi.org/10.1093/ve/veaf104
-
[15]
Matthew R. Henn et al. “Whole Genome Deep Sequencing of HIV-1 Reveals the Impact of Early Minor Variants Upon Immune Recognition During Acute Infection”. In:PLOS Pathogens8.3 (Mar. 2012), pp. 1–14.doi:10.1371/journal.ppat.1002529.url:https://doi.org/10. 1371/journal.ppat.1002529
work page doi:10.1371/journal.ppat.1002529.url:https://doi.org/10 2012
-
[16]
Rapid Evolution of RNA Genomes
John Holland et al. “Rapid Evolution of RNA Genomes”. In:Science215.4540 (1982), pp. 1577– 1585.doi:10.1126/science.7041255. eprint:https://www.science.org/doi/pdf/10. 1126/science.7041255.url:https://www.science.org/doi/abs/10.1126/science. 7041255
-
[17]
Breaking records in the evolutionary race
Joachim Krug and Kavita Jain. “Breaking records in the evolutionary race”. In:Physica A: Sta- tistical Mechanics and its Applications358.1 (2005). Condensed Matter and Statistical Physics, pp. 1–9.issn: 0378-4371.doi:https://doi.org/10.1016/j.physa.2005.06.002.url: https://www.sciencedirect.com/science/article/pii/S0378437105005753
-
[18]
Quasispecies Theory and the Behavior of RNA Viruses
Adam S. Lauring and Raul Andino. “Quasispecies Theory and the Behavior of RNA Viruses”. In:PLOS Pathogens6.7 (July 2010), pp. 1–8.doi:10.1371/journal.ppat.1001005.url: https://doi.org/10.1371/journal.ppat.1001005. 17
-
[19]
SARS-CoV-2 within-host diversity and transmission
Katrina A. Lythgoe et al. “SARS-CoV-2 within-host diversity and transmission”. In:Science 372.6539 (2021), eabg0821.doi:10.1126/science.abg0821. eprint:https://www.science. org/doi/pdf/10.1126/science.abg0821.url:https://www.science.org/doi/abs/10. 1126/science.abg0821
-
[20]
Susanna Manrubia, Luis F. Seoane, and Jos´ e A. Cuesta. “The challenge of scale in molecular adaptaton: Local searches in astronomical genotype networks”. In:Integrative Theory of Evo- lution – Aspects and Insights. Ed. by Guenther Witzany. Springer, 2026.doi:10.1007/978- 3-032-24295-2_26
-
[21]
M Martell et al. “Hepatitis C virus (HCV) circulates as a population of different but closely related genomes: quasispecies nature of HCV genome distribution”. In:Journal of Virology 66.5 (1992), pp. 3225–3229.doi:10 . 1128 / jvi . 66 . 5 . 3225 - 3229 . 1992. eprint:https : / / journals . asm . org / doi / pdf / 10 . 1128 / jvi . 66 . 5 . 3225 - 3229 . 1...
-
[22]
Brenda Mart´ ınez-Gonz´ alez et al. “SARS-CoV-2 Mutant Spectra at Different Depth Levels Reveal an Overwhelming Abundance of Low Frequency Mutations”. In:Pathogens11.6 (2022). issn: 2076-0817.doi:10.3390/pathogens11060662.url:https://www.mdpi.com/2076- 0817/11/6/662
work page doi:10.3390/pathogens11060662.url:https://www.mdpi.com/2076- 2022
-
[23]
SARS-CoV-2 mutant spectrum complexity is an epidemio- logically evolvable trait
Brenda Mart´ ınez-Gonz´ alez et al. “SARS-CoV-2 mutant spectrum complexity is an epidemio- logically evolvable trait”. In:Proceedings of the National Academy of Sciences122.39 (2025), e2515706122.doi:10.1073/pnas.2515706122. eprint:https://www.pnas.org/doi/pdf/10. 1073/pnas.2515706122.url:https://www.pnas.org/doi/abs/10.1073/pnas.2515706122
-
[24]
SARS-CoV-2 Point Mutation and Deletion Spectra and Their Association with Different Disease Outcomes
Brenda Mart´ ınez-Gonz´ alez et al. “SARS-CoV-2 Point Mutation and Deletion Spectra and Their Association with Different Disease Outcomes”. In:Microbiology Spectrum10.2 (2022), e00221– 22.doi:10.1128/spectrum.00221-22. eprint:https://journals.asm.org/doi/pdf/10. 1128/spectrum.00221-22.url:https://journals.asm.org/doi/abs/10.1128/spectrum. 00221-22
-
[25]
Increased RNA virus population diversity improves adaptability
Florian Mattenberger, Marina Vila-Nistal, and Ron Geller. “Increased RNA virus population diversity improves adaptability”. In:Scientific Reports11.1 (Mar. 2021), p. 6824.issn: 2045- 2322.doi:10.1038/s41598-021-86375-z.url:https://doi.org/10.1038/s41598-021- 86375-z
work page doi:10.1038/s41598-021-86375-z.url:https://doi.org/10.1038/s41598-021- 2021
-
[26]
Structure and dynamics of SARS-CoV-2 proofreading exoribonu- clease ExoN
Nicholas H. Moeller et al. “Structure and dynamics of SARS-CoV-2 proofreading exoribonu- clease ExoN”. In:Proceedings of the National Academy of Sciences119.9 (2022), e2106379119. doi:10.1073/pnas.2106379119.url:https://www.pnas.org/doi/abs/10.1073/pnas. 2106379119
work page doi:10.1073/pnas.2106379119.url:https://www.pnas.org/doi/abs/10.1073/pnas 2022
-
[27]
Assortative mixing in networks
M. E. J. Newman. “Assortative mixing in networks”. In:Phys. Rev. Lett.89 (2002), p. 208701
2002
-
[28]
M. E. J. Newman.Networks: An introduction. Oxford University Press, New York, 2010
2010
-
[29]
High-throughput sequencing (HTS) for the analysis of viral pop- ulations
Marcos P´ erez-Losada et al. “High-throughput sequencing (HTS) for the analysis of viral pop- ulations”. In:Infection, Genetics and Evolution80 (2020), p. 104208.issn: 1567-1348.doi: https://doi.org/10.1016/j.meegid.2020.104208.url:https://www.sciencedirect. com/science/article/pii/S156713482030040X
work page doi:10.1016/j.meegid.2020.104208.url:https://www.sciencedirect 2020
-
[30]
Recent advances in inferring viral diversity from high-throughput sequ encing data
Susana Posada-Cespedes, David Seifert, and Niko Beerenwinkel. “Recent advances in inferring viral diversity from high-throughput sequ encing data”. In:Virus Research239 (2017). Deep sequencing in virology, pp. 17–32.issn: 0168-1702.doi:https : / / doi . org / 10 . 1016 / j . virusres.2016.09.016.url:https://www.sciencedirect.com/science/article/pii/ S0168...
2017
-
[31]
Deep sequencing in the management of hepatitis virus infections
Josep Quer et al. “Deep sequencing in the management of hepatitis virus infections”. In:Virus Research239 (2017). Deep sequencing in virology, pp. 115–125.issn: 0168-1702.doi:https: //doi.org/10.1016/j.virusres.2016.12.020.url:https://www.sciencedirect.com/ science/article/pii/S0168170216304567. 18
work page doi:10.1016/j.virusres.2016.12.020.url:https://www.sciencedirect.com/ 2017
-
[32]
Growth and Structural Transformation
Rafael Sanju´ an and Pilar Domingo-Calap. “Genetic Diversity and Evolution of Viral Popula- tions”. In:Encyclopedia of Virology (Fourth Edition). Ed. by Dennis H. Bamford and Mark Zuckerman. Fourth Edition. Oxford: Academic Press, 2021, pp. 53–61.isbn: 978-0-12-814516- 6.doi:https://doi.org/10.1016/B978- 0- 12- 809633- 8.20958- 8.url:https://www. scienced...
-
[33]
The total number and mass of SARS-CoV-2 virions
Ron Sender et al. “The total number and mass of SARS-CoV-2 virions”. In:Proceedings of the National Academy of Sciences118.25 (2021), e2024815118.doi:10.1073/pnas.2024815118. eprint:https://www.pnas.org/doi/pdf/10.1073/pnas.2024815118.url:https://www. pnas.org/doi/abs/10.1073/pnas.2024815118
-
[34]
Hierarchical genotype networks and incipient ecological speciation in Qβphage quasispecies
Lu´ ıs F. Seoane et al. “Hierarchical genotype networks and incipient ecological speciation in Qβphage quasispecies”. In:Proceedings of the National Academy of Sciences123.14 (2026), e2512150123.doi:10.1073/pnas.2512150123. eprint:https://www.pnas.org/doi/pdf/10. 1073/pnas.2512150123.url:https://www.pnas.org/doi/abs/10.1073/pnas.2512150123
-
[35]
Pilar Somovilla, Susanna Manrubia, and Ester L´ azaro. “Evolutionary Dynamics in the RNA Bacteriophage QβDepends on the Pattern of Change in Selective Pressures”. In:Pathogens 8.2 (2019).doi:10.3390/pathogens8020080
-
[36]
Standing Genetic Diversity and Transmission Bottleneck Size Drive Adaptation in Bacteriophage Qβ
Pilar Somovilla et al. “Standing Genetic Diversity and Transmission Bottleneck Size Drive Adaptation in Bacteriophage Qβ”. In:International Journal of Molecular Sciences23.16 (2022). doi:10.3390/ijms23168876
-
[37]
The mutational landscape of SARS-CoV-2 provides new insight into viral evolution and fitness
Jori Symons et al. “The mutational landscape of SARS-CoV-2 provides new insight into viral evolution and fitness”. In:Nature Communications16.1 (July 2025), p. 6425.issn: 2041-1723. doi:10.1038/s41467-025-61555-x.url:https://doi.org/10.1038/s41467-025-61555- x
work page doi:10.1038/s41467-025-61555-x.url:https://doi.org/10.1038/s41467-025-61555- 2025
-
[38]
Genotype-to-Protein Map and Collective Adaptation in a Viral Pop- ulation
Ariadna Villanueva et al. “Genotype-to-Protein Map and Collective Adaptation in a Viral Pop- ulation”. In:Biophysica2.4 (2022), pp. 381–399.issn: 2673-4125.doi:10.3390/biophysica2040034. url:https://www.mdpi.com/2673-4125/2/4/34
-
[39]
Immunity-induced criticality of the genotype network of influenza A (H3N2) hemagglutinin
Blake J M Williams et al. “Immunity-induced criticality of the genotype network of influenza A (H3N2) hemagglutinin”. In:PNAS Nexus1.4 (Aug. 2022), pgac143.doi:10.1093/pnasnexus/ pgac143. eprint:https : / / academic . oup . com / pnasnexus / article - pdf / 1 / 4 / pgac143 / 48849248/pgac143.pdf.url:https://doi.org/10.1093/pnasnexus/pgac143. 19 Supplement...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.