Gaussian Mixture Model-Based Focused Refinement for Enhanced Flexible Structure Determination in CryoEM and CryoET
Pith reviewed 2026-06-28 23:27 UTC · model grok-4.3
The pith
A Gaussian mixture model-based focused alignment procedure improves resolution of small domains in dynamic proteins for both CryoEM and in situ CryoET.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
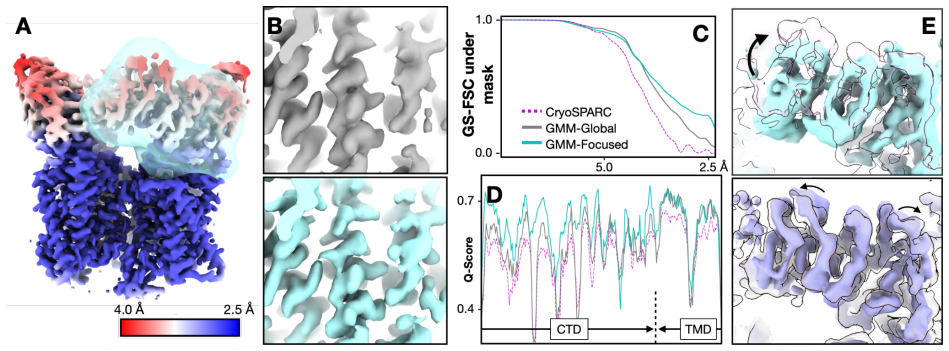
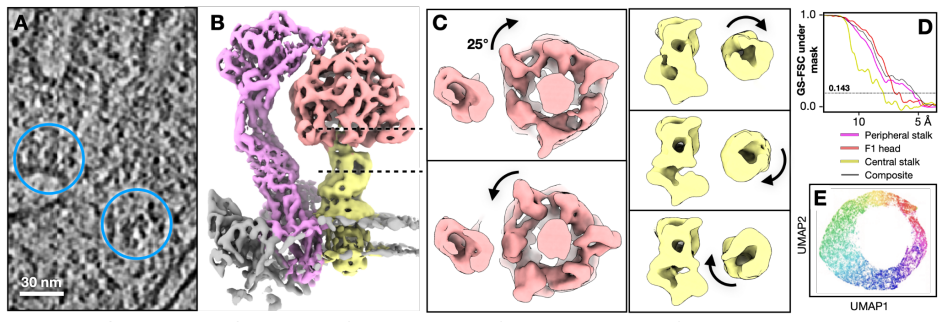
The central claim is that a unified refinement pipeline using a Gaussian mixture model-based focused alignment procedure enhances flexible protein structure determination in both CryoEM and in situ CryoET by improving resolution of small domains in highly dynamic proteins and revealing intricate conformational changes, as demonstrated by correcting the per-subunit motion of TRPV1 and capturing the rotary dynamics of ATP synthase within mitochondria.
What carries the argument
Gaussian mixture model-based focused alignment procedure, which models density to focus refinement and correct per-subunit motions.
If this is right
- Resolution of small domains in highly dynamic proteins is improved.
- Intricate conformational changes are revealed in the structures.
- Per-subunit motion of TRPV1 is corrected.
- Rotary dynamics of ATP synthase within mitochondria are captured.
- The same pipeline applies to both CryoEM and in situ CryoET data.
Where Pith is reading between the lines
- The approach could extend to other flexible macromolecular complexes beyond the two examples shown.
- Better resolved dynamic structures might aid interpretation of how conformational changes drive function.
- Testing on additional independent datasets would clarify how general the motion-correction benefit is.
Load-bearing premise
The Gaussian mixture model accurately represents and corrects per-subunit motions in highly flexible proteins without introducing alignment artifacts or biasing the resulting density maps.
What would settle it
If refined density maps of TRPV1 or ATP synthase show no resolution gain or contain visible artifacts compared to standard refinement, the central claim would be falsified.
Figures
read the original abstract
Dynamic conformational changes of proteins are crucial for their cellular functions. Here we present a unified refinement pipeline for flexible protein structures in both CryoEM and in situ CryoET. Using a Gaussian mixture model-based focused alignment procedure, we improve resolution of small domains in highly dynamic proteins and reveal intricate conformational changes. The method corrects the per-subunit motion of TRPV1 and captures the rotary dynamics of ATP synthase within mitochondria.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents a unified refinement pipeline for flexible protein structures in CryoEM and CryoET that employs a Gaussian mixture model-based focused alignment procedure. The central claims are that this approach improves resolution of small domains in highly dynamic proteins, corrects per-subunit motion in TRPV1, and captures the rotary dynamics of ATP synthase within mitochondria while revealing intricate conformational changes.
Significance. If the GMM-based focused alignment demonstrably improves resolution and captures dynamics without introducing artifacts, the method would provide a practical advance for handling flexibility in both isolated complexes and in situ cellular environments. The unified treatment of CryoEM and CryoET is a useful framing, though the absence of quantitative validation metrics limits assessment of its broader impact.
major comments (2)
- [Abstract] Abstract: the claims of resolution improvement and motion correction for TRPV1 and ATP synthase are presented without any reported resolution values, FSC curves, particle numbers, or comparison to standard refinement, preventing evaluation of whether the GMM procedure actually delivers the stated gains.
- [Abstract] Abstract: the weakest assumption—that the GMM accurately models per-subunit motions without biasing the density or creating alignment artifacts—remains untested in the visible text; no cross-validation, simulated-data tests, or artifact checks are described.
minor comments (1)
- [Abstract] The abstract uses both 'focused alignment procedure' and 'focused refinement'; consistent terminology would improve clarity.
Simulated Author's Rebuttal
We thank the referee for their comments on our manuscript. We address each major comment below and will make revisions to improve the clarity and validation of our method.
read point-by-point responses
-
Referee: [Abstract] Abstract: the claims of resolution improvement and motion correction for TRPV1 and ATP synthase are presented without any reported resolution values, FSC curves, particle numbers, or comparison to standard refinement, preventing evaluation of whether the GMM procedure actually delivers the stated gains.
Authors: The abstract provides a high-level summary of the results. However, we agree that including specific quantitative details would aid evaluation. In the revised version, we will incorporate key resolution values, references to FSC curves, particle numbers, and comparisons to standard methods directly into the abstract. These metrics are detailed in the main text and supplementary materials for both the TRPV1 and ATP synthase examples. revision: yes
-
Referee: [Abstract] Abstract: the weakest assumption—that the GMM accurately models per-subunit motions without biasing the density or creating alignment artifacts—remains untested in the visible text; no cross-validation, simulated-data tests, or artifact checks are described.
Authors: We recognize the importance of validating the core assumptions of the GMM approach. While the manuscript describes the method and its application, we agree that more explicit validation is needed. We will revise the manuscript to include descriptions of cross-validation procedures, tests on simulated data, and checks for potential artifacts to demonstrate that the GMM modeling does not bias the density or introduce alignment issues. revision: yes
Circularity Check
No derivation chain or equations presented; circularity cannot be identified
full rationale
The provided abstract and placeholder full text contain no equations, fitting procedures, derivation steps, or self-citations that could be inspected for circularity. The method is described at a high level as a 'Gaussian mixture model-based focused alignment procedure' without any mathematical formulation, parameter fitting details, or claimed predictions that reduce to inputs. Per the rules, circularity requires explicit quotes exhibiting reduction by construction; none exist here. This is the normal case of a paper whose technical content is not visible for analysis, so the score is 0 with no steps.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Gilles, M. A. & Singer, A. Cryo-EM heterogeneity analysis using regularized covariance estimation and kernel regression. Proc. Natl. Acad. Sci. U. S. A. 122, e2419140122 (2025). 13. Schwab, J., Kimanius, D., Burt, A., Dendooven, T. & Scheres, S. H. W. DynaMight: estimating molecular motions with improved reconstruction from cryo-EM images. Nat. Methods 21...
-
[2]
Chen, M., Toader, B. & Lederman, R. Integrating Molecular Models Into CryoEM Heterogeneity Analysis Using Scalable High-resolution Deep Gaussian Mixture Models. J. Mol. Biol. 168014 (2023). 25. Harris, J. A. et al. Selective G protein signaling driven by substance P-neurokinin receptor dynamics. Nat. Chem. Biol. 18, 109–115 (2022). 26. Croll, T. I. ISOLDE...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.