TrioSeq: A Novel Approach to Accelerate Triplet Sequence Alignment on GPUs
Pith reviewed 2026-06-29 09:52 UTC · model grok-4.3
The pith
TrioSeq implements 3-way sequence alignment on GPUs using new parallelism and synchronization levels, outperforming prior GPU methods by at least 20% on simulated genomic data.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
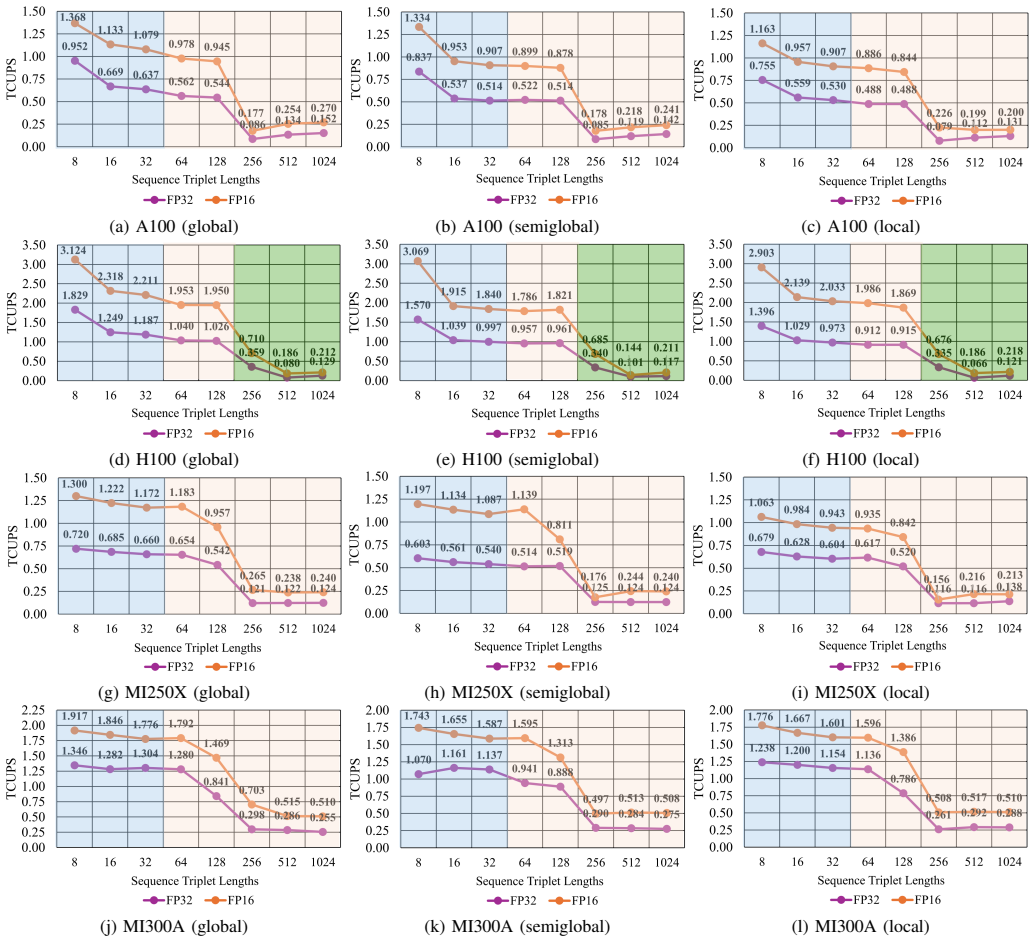
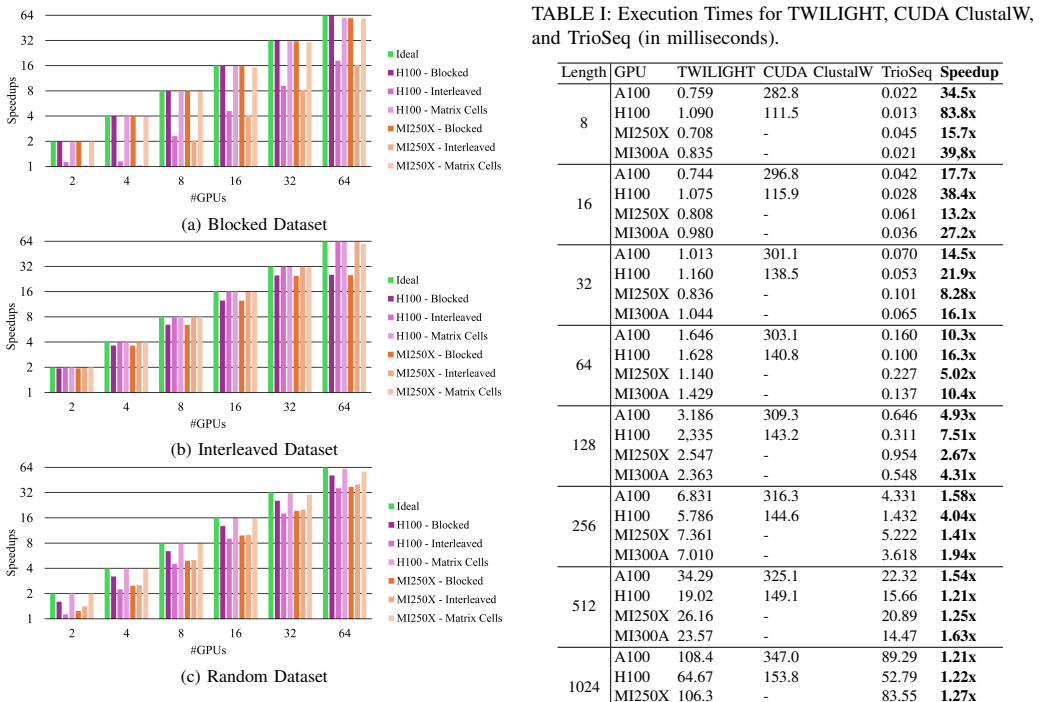
TrioSeq is proposed as a fine-grained strategy to efficiently implement 3-way alignments on GPUs, leveraging novel levels of GPU parallelism and synchronization to achieve high throughput in aligning sequence triplets. Evaluation on NVIDIA and AMD GPUs shows that TrioSeq outperforms state-of-the-art GPU progressive methods on 3-way alignment by at least 20% on simulated genomic datasets.
What carries the argument
The fine-grained strategy for 3-way alignments that exploits novel GPU parallelism and synchronization levels to raise throughput for sequence triplets.
If this is right
- Exact 3-way alignments become feasible as the first step in progressive MSA pipelines.
- Genomic analysis pipelines gain speed on both NVIDIA and AMD hardware without changing the alignment algorithm.
- Higher throughput for triplets supports scaling MSA to larger numbers of sequences.
- The same parallelism approach can be applied to other exact k-way alignment problems.
Where Pith is reading between the lines
- Adoption could reduce total compute time for large genome projects that currently start from pairwise alignments.
- The method opens a path to test whether triplet-based trees improve downstream phylogenetic accuracy on real data.
- Hardware vendors may add explicit support for the cross-thread intrinsics used here once the pattern is public.
Load-bearing premise
The performance advantage measured on simulated datasets will generalize to real genomic sequences and the implementation can be integrated into existing MSA workflows without hidden costs or accuracy trade-offs.
What would settle it
Measure wall-clock time and output quality when running TrioSeq on a collection of real genomic sequences and compare directly against current GPU 3-way methods, or test full integration into an MSA tool and record any added overhead.
Figures

read the original abstract
State-of-the-art multiple sequence alignment (MSA) algorithms are based on progressive approaches that rely on pairwise sequence alignment (PSA) to generate guide trees to align all sequences. Given an evidenced explosion in genomic data availability, research efforts have focused on accelerating PSA on massively-parallel architectures (e.g., GPUs) and specialized hardware (e.g., FPGAs). However, there is increasing evidence that starting from exact 3-way alignments could provide more robust, accurate MSAs, and improve genomic analysis. While the current literature has shown that PSA algorithms can be extended to align sequence triplets, the existent state-of-the-art on hardware acceleration of exact 3-way alignments is still scarce. In particular, current GPU methods are still inefficient due to lacking support for novel hardware features (e.g., cross-thread intrinsics), while being closed-source and vendor-specific. In this paper, TrioSeq is proposed as a fine-grained strategy to efficiently implement 3-way alignments on GPUs, leveraging novel levels of GPU parallelism and synchronization to achieve high throughput in aligning sequence triplets. Evaluation on NVIDIA and AMD GPUs shows that TrioSeq outperforms state-of-the-art GPU progressive methods on 3-way alignment by at least 20% on simulated genomic datasets.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper proposes TrioSeq, a fine-grained GPU strategy for exact 3-way sequence alignment that exploits novel levels of parallelism and synchronization (including cross-thread intrinsics). It reports that this approach outperforms state-of-the-art GPU progressive methods on 3-way alignment by at least 20% when evaluated on simulated genomic datasets using both NVIDIA and AMD hardware.
Significance. A verified, cross-vendor GPU implementation that delivers measurable throughput gains on triplet alignment could support more accurate progressive MSA pipelines if accuracy is preserved. The emphasis on open, non-vendor-specific code and hardware-feature utilization is a positive contribution if the empirical claims are substantiated with reproducible benchmarks.
major comments (3)
- [Evaluation] Evaluation section: the central performance claim (≥20% speedup) is reported exclusively on simulated genomic datasets; no timings or accuracy results on real genomic sequences are supplied, leaving open whether the advantage persists under realistic length distributions, substitution patterns, and gap structures.
- [Evaluation] Evaluation section: no alignment-quality metrics (sum-of-pairs score, column score, or SP score) or statistical error analysis are provided to demonstrate that the reported throughput gain does not trade off against alignment fidelity relative to the GPU progressive baselines.
- [Abstract] Abstract and Evaluation section: the manuscript supplies no benchmark specifications, dataset parameters, number of replicates, or variance measures, rendering the 20% improvement claim impossible to assess for statistical robustness or reproducibility.
minor comments (1)
- The claim that current GPU methods lack support for 'novel hardware features (e.g., cross-thread intrinsics)' would benefit from a precise citation to the specific intrinsics newly exploited by TrioSeq versus prior work.
Simulated Author's Rebuttal
We thank the referee for the constructive comments on the evaluation of TrioSeq. We address each major comment below and indicate the revisions we will make.
read point-by-point responses
-
Referee: [Evaluation] Evaluation section: the central performance claim (≥20% speedup) is reported exclusively on simulated genomic datasets; no timings or accuracy results on real genomic sequences are supplied, leaving open whether the advantage persists under realistic length distributions, substitution patterns, and gap structures.
Authors: Simulated datasets were selected to enable controlled variation of sequence lengths, substitution rates, and gap structures while providing ground-truth alignments for verification. This is a common practice for isolating algorithmic performance in the MSA literature. We agree that real-sequence results would strengthen the claims and will add timing experiments on real genomic data (e.g., from NCBI RefSeq) in the revised manuscript. revision: yes
-
Referee: [Evaluation] Evaluation section: no alignment-quality metrics (sum-of-pairs score, column score, or SP score) or statistical error analysis are provided to demonstrate that the reported throughput gain does not trade off against alignment fidelity relative to the GPU progressive baselines.
Authors: TrioSeq computes exact 3-way alignments via the same dynamic-programming recurrence used by the baselines; therefore alignment fidelity is identical by construction and no accuracy–speed trade-off exists. Nevertheless, to address the concern explicitly, the revised Evaluation section will report sum-of-pairs and column scores together with statistical comparisons confirming equivalence. revision: yes
-
Referee: [Abstract] Abstract and Evaluation section: the manuscript supplies no benchmark specifications, dataset parameters, number of replicates, or variance measures, rendering the 20% improvement claim impossible to assess for statistical robustness or reproducibility.
Authors: We acknowledge that these experimental details were omitted. The revised manuscript will expand the Evaluation section with full benchmark specifications, including dataset parameters, the number of replicates performed, and variance measures (mean and standard deviation) to support reproducibility of the reported gains. revision: yes
Circularity Check
No circularity: empirical performance claim only
full rationale
The paper proposes a GPU implementation (TrioSeq) for exact 3-way sequence alignment and reports measured throughput on simulated genomic datasets. The abstract and provided text contain no equations, derivations, fitted parameters, predictions, or first-principles results. The central claim is a direct empirical comparison (≥20% speedup vs. prior GPU methods), with no self-definitional steps, fitted-input-as-prediction, or load-bearing self-citations that reduce the result to its own inputs by construction. The derivation chain is absent; the work is self-contained as an engineering/implementation paper.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Next-generation sequencing technologies: An overview,
T. Hu, N. Chitnis, D. Monos, and A. Dinh, “Next-generation sequencing technologies: An overview,”Human immunology, vol. 82, no. 11, pp. 801–811, 2021
2021
-
[2]
A global reference for human genetic variation,
G. P. Consortiumet al., “A global reference for human genetic variation,”Nature, vol. 526, no. 7571, p. 68, 2015
2015
-
[3]
Giab: Genome reference material development resources for clinical sequencing,
C. Xiao, J. Zook, S. Trask, S. Sherry, and G. in-a Bottle Consortium, “Giab: Genome reference material development resources for clinical sequencing,”Cancer Research, vol. 74, no. 19 Supplement, pp. 5328– 5328, 2014
2014
-
[4]
Petabase- scale sequence alignment catalyses viral discovery,
R. C. Edgar, B. Taylor, V . Lin, T. Altman, P. Barbera, D. Meleshko, D. Lohr, G. Novakovsky, B. Buchfink, B. Al-Shayebet al., “Petabase- scale sequence alignment catalyses viral discovery,”Nature, vol. 602, no. 7895, pp. 142–147, 2022
2022
-
[5]
Multiple sequence alignment accuracy and phylogenetic inference,
T. H. Ogden and M. S. Rosenberg, “Multiple sequence alignment accuracy and phylogenetic inference,”Systematic biology, vol. 55, no. 2, pp. 314–328, 2006
2006
-
[6]
A comparative as- sessment and analysis of 20 representative sequence alignment methods for protein structure prediction,
R. Yan, D. Xu, J. Yang, S. Walker, and Y . Zhang, “A comparative as- sessment and analysis of 20 representative sequence alignment methods for protein structure prediction,”Scientific reports, vol. 3, no. 1, p. 2619, 2013
2013
-
[7]
A survey on sequence alignment algorithms and state-of-the-art aligners,
K. Prousalis, K. Georgiou, A. Kalogeropoulos, D. Ntalaperas, N. Kono- faos, L. Aggelis, C. Papalitsas, T. Stavropoulos, and N. Gariboldi, “A survey on sequence alignment algorithms and state-of-the-art aligners,” ACM Computing Surveys, 2025
2025
-
[8]
A general method applicable to the search for similarities in the amino acid sequence of two proteins,
S. B. Needleman and C. D. Wunsch, “A general method applicable to the search for similarities in the amino acid sequence of two proteins,” Journal of molecular biology, vol. 48, no. 3, pp. 443–453, 1970
1970
-
[9]
Identification of common molecular subsequences,
T. F. Smith, M. S. Watermanet al., “Identification of common molecular subsequences,”Journal of molecular biology, vol. 147, no. 1, pp. 195– 197, 1981
1981
-
[10]
Computational complexity of multiple sequence alignment with sp-score,
W. Just, “Computational complexity of multiple sequence alignment with sp-score,”Journal of computational biology, vol. 8, no. 6, pp. 615– 623, 2001
2001
-
[11]
The multiple sequence alignment problem in biology,
H. Carrillo and D. Lipman, “The multiple sequence alignment problem in biology,”SIAM journal on applied mathematics, vol. 48, no. 5, pp. 1073–1082, 1988
1988
-
[12]
Progressive multiple sequence alignment with indel evolution,
M. Maiolo, X. Zhang, M. Gil, and M. Anisimova, “Progressive multiple sequence alignment with indel evolution,”BMC bioinformatics, vol. 19, no. 1, p. 331, 2018
2018
-
[13]
Clustal w: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice,
J. D. Thompson, D. G. Higgins, and T. J. Gibson, “Clustal w: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice,”Nucleic acids research, vol. 22, no. 22, pp. 4673–4680, 1994
1994
-
[14]
Construction of phylogenetic tree using upgma method,
A. Kaur and M. Kaur, “Construction of phylogenetic tree using upgma method,” in2024 7th International Conference on Contemporary Com- puting and Informatics (IC3I), vol. 7. IEEE, 2024, pp. 242–246
2024
-
[15]
Construction of phylogenetic tree using neighbor joining algorithms to identify the host and the spreading of sars epidemic,
M. I. Irawan and S. Amiroch, “Construction of phylogenetic tree using neighbor joining algorithms to identify the host and the spreading of sars epidemic,”Journal of Theoretical and Applied Information Technology, vol. 71, no. 3, pp. 424–429, 2015
2015
-
[16]
Recent evolutions of multiple sequence alignment algorithms,
C. Notredame, “Recent evolutions of multiple sequence alignment algorithms,”PLoS computational biology, vol. 3, no. 8, p. e123, 2007
2007
-
[17]
M- coffee: combining multiple sequence alignment methods with t-coffee,
I. M. Wallace, O. O’sullivan, D. G. Higgins, and C. Notredame, “M- coffee: combining multiple sequence alignment methods with t-coffee,” Nucleic acids research, vol. 34, no. 6, pp. 1692–1699, 2006
2006
-
[18]
Correlations between alignment gaps and nucleotide substitution or amino acid replacement,
T.-K. Seo, B. D. Redelings, and J. L. Thorne, “Correlations between alignment gaps and nucleotide substitution or amino acid replacement,” Proceedings of the National Academy of Sciences, vol. 119, no. 34, p. e2204435119, 2022
2022
-
[19]
Phylogeny-aware gap placement pre- vents errors in sequence alignment and evolutionary analysis,
A. Loytynoja and N. Goldman, “Phylogeny-aware gap placement pre- vents errors in sequence alignment and evolutionary analysis,”science, vol. 320, no. 5883, pp. 1632–1635, 2008
2008
-
[20]
Progressive multiple sequence alignments from triplets,
M. Kruspe and P. F. Stadler, “Progressive multiple sequence alignments from triplets,”BMC bioinformatics, vol. 8, no. 1, p. 254, 2007
2007
-
[21]
Multiple sequence alignment accuracy and evolu- tionary distance estimation,
M. S. Rosenberg, “Multiple sequence alignment accuracy and evolu- tionary distance estimation,”Bmc Bioinformatics, vol. 6, no. 1, p. 278, 2005
2005
-
[22]
Lower bounds on multiple sequence alignment using exact 3-way alignment,
C. J. Colbourn and S. Kumar, “Lower bounds on multiple sequence alignment using exact 3-way alignment,”BMC bioinformatics, vol. 8, no. 1, p. 140, 2007
2007
-
[23]
Three-way alignment improves multiple sequence alignment of highly diverged sequences,
M. Askari Rad, A. Kruglikov, and X. Xia, “Three-way alignment improves multiple sequence alignment of highly diverged sequences,” Algorithms, vol. 17, no. 5, p. 205, 2024
2024
-
[24]
Measuring guide-tree depen- dency of inferred gaps in progressive aligners,
S. Capella-Guti ´errez and T. Gabald ´on, “Measuring guide-tree depen- dency of inferred gaps in progressive aligners,”Bioinformatics, vol. 29, no. 8, pp. 1011–1017, 2013
2013
-
[25]
Major revisions in arthropod phylogeny through improved supermatrix, with support for two possible waves of land invasion by chelicerates,
K. E. Noah, J. Hao, L. Li, X. Sun, B. Foley, Q. Yang, and X. Xia, “Major revisions in arthropod phylogeny through improved supermatrix, with support for two possible waves of land invasion by chelicerates,” Evolutionary Bioinformatics, vol. 16, p. 1176934320903735, 2020
2020
-
[26]
Impact of alignment algorithm on the estimation of pair- wise genetic similarity of porcine reproductive and respiratory syndrome virus (prrsv),
M.- `E. Lambert, J. Arsenault, B. Delisle, P. Audet, Z. Poljak, and S. D’Allaire, “Impact of alignment algorithm on the estimation of pair- wise genetic similarity of porcine reproductive and respiratory syndrome virus (prrsv),”BMC Veterinary Research, vol. 15, no. 1, p. 135, 2019
2019
-
[27]
Accurate consistency- based msa reducing the memory footprint,
J. Llad ´os, F. Cores, F. Guirado, and J. L. L ´erida, “Accurate consistency- based msa reducing the memory footprint,”Computer Methods and Programs in Biomedicine, vol. 208, p. 106237, 2021
2021
-
[28]
Alignment of three biological sequences with an efficient traceback procedure,
O. Gotoh, “Alignment of three biological sequences with an efficient traceback procedure,”Journal of Theoretical Biology, vol. 121, no. 3, pp. 327–337, 1986
1986
-
[29]
Browniealigner: accurate alignment of illumina sequencing data to de bruijn graphs,
M. Heydari, G. Miclotte, Y . Van de Peer, and J. Fostier, “Browniealigner: accurate alignment of illumina sequencing data to de bruijn graphs,” BMC bioinformatics, vol. 19, no. 1, p. 311, 2018
2018
-
[30]
Parasail: Simd c library for global, semi-global, and local pairwise sequence alignments,
J. Daily, “Parasail: Simd c library for global, semi-global, and local pairwise sequence alignments,”BMC bioinformatics, vol. 17, no. 1, p. 81, 2016
2016
-
[31]
Performance extraction and suitability analysis of multi-and many-core architectures for next generation sequencing secondary analysis,
S. Misra, T. C. Pan, K. Mahadik, G. Powley, P. N. Vaidya, M. Vasimud- din, and S. Aluru, “Performance extraction and suitability analysis of multi-and many-core architectures for next generation sequencing secondary analysis,” inProceedings of the 27th International Conference on Parallel Architectures and Compilation Techniques, 2018, pp. 1–14
2018
-
[32]
Generic accelerated sequence alignment in seqan using vectoriza- tion and multi-threading,
R. Rahn, S. Budach, P. Costanza, M. Ehrhardt, J. Hancox, and K. Rein- ert, “Generic accelerated sequence alignment in seqan using vectoriza- tion and multi-threading,”Bioinformatics, vol. 34, no. 20, pp. 3437– 3445, 2018
2018
-
[33]
Cudalign 4.0: Incremental speculative traceback for exact chromosome-wide alignment in gpu clusters,
E. F. de Oliveira Sandes, G. Miranda, X. Martorell, E. Ayguade, G. Teodoro, and A. C. M. Melo, “Cudalign 4.0: Incremental speculative traceback for exact chromosome-wide alignment in gpu clusters,”IEEE Transactions on Parallel and Distributed Systems, vol. 27, no. 10, pp. 2838–2850, 2016
2016
-
[34]
Any- seq/gpu: a novel approach for faster sequence alignment on gpus,
A. M ¨uller, B. Schmidt, R. Membarth, R. Leißa, and S. Hack, “Any- seq/gpu: a novel approach for faster sequence alignment on gpus,” in Proceedings of the 36th ACM International Conference on Supercom- puting, 2022, pp. 1–11
2022
-
[35]
Gasal2: a gpu accelerated sequence alignment library for high- throughput ngs data,
N. Ahmed, J. L ´evy, S. Ren, H. Mushtaq, K. Bertels, and Z. Al- Ars, “Gasal2: a gpu accelerated sequence alignment library for high- throughput ngs data,”BMC bioinformatics, vol. 20, no. 1, p. 520, 2019
2019
-
[36]
Adept: a domain independent sequence alignment strategy for gpu architectures,
M. G. Awan, J. Deslippe, A. Buluc, O. Selvitopi, S. Hofmeyr, L. Oliker, and K. Yelick, “Adept: a domain independent sequence alignment strategy for gpu architectures,”BMC bioinformatics, vol. 21, no. 1, p. 406, 2020
2020
-
[37]
Cudasw++ 4.0: ultra-fast gpu-based smith–waterman protein sequence database search,
B. Schmidt, F. Kallenborn, A. Chacon, and C. Hundt, “Cudasw++ 4.0: ultra-fast gpu-based smith–waterman protein sequence database search,” BMC bioinformatics, vol. 25, no. 1, p. 342, 2024
2024
-
[38]
Space efficient sequence alignment for sram-based computing: X-drop on the graphcore ipu,
L. Burchard, M. X. Zhao, J. Langguth, A. Buluc ¸, and G. Guidi, “Space efficient sequence alignment for sram-based computing: X-drop on the graphcore ipu,” inProceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis, 2023, pp. 1–16
2023
-
[39]
ipuma: High-performance sequence alignment on the graphcore ipu,
M. Zhao, L. Burchard, D. T. Schroeder, J. Langguth, and X. Cai, “ipuma: High-performance sequence alignment on the graphcore ipu,” inISC High Performance 2024 Research Paper Proceedings (39th International Conference). Prometeus GmbH, 2024, pp. 1–11
2024
-
[40]
Fpgasw: acceler- ating large-scale smith–waterman sequence alignment application with backtracking on fpga linear systolic array,
X. Fei, Z. Dan, L. Lina, M. Xin, and Z. Chunlei, “Fpgasw: acceler- ating large-scale smith–waterman sequence alignment application with backtracking on fpga linear systolic array,”Interdisciplinary Sciences: Computational Life Sciences, vol. 10, no. 1, pp. 176–188, 2018
2018
-
[41]
An fpga accelerator of the wavefront algorithm for genomics pairwise alignment,
A. Haghi, S. Marco-Sola, L. Alvarez, D. Diamantopoulos, C. Hagleitner, and M. Moreto, “An fpga accelerator of the wavefront algorithm for genomics pairwise alignment,” in2021 31st International Conference on Field-Programmable Logic and Applications (FPL). IEEE, 2021, pp. 151–159
2021
-
[42]
Swifold: Smith-waterman implementation on fpga with opencl for long dna sequences,
E. Rucci, C. Garcia, G. Botella, A. De Giusti, M. Naiouf, and M. Prieto- Matias, “Swifold: Smith-waterman implementation on fpga with opencl for long dna sequences,”BMC systems biology, vol. 12, no. Suppl 5, p. 96, 2018
2018
-
[43]
A block-based systolic array on an hbm2 fpga for dna sequence alignment,
R. Ben Abdelhamid and Y . Yamaguchi, “A block-based systolic array on an hbm2 fpga for dna sequence alignment,” inInternational Symposium on Applied Reconfigurable Computing. Springer, 2020, pp. 298–313
2020
-
[44]
Accelerating multiple sequence alignments using parallel computing,
Q. Bani Baker, R. A. Al-Hussien, and M. Al-Ayyoub, “Accelerating multiple sequence alignments using parallel computing,”Computation, vol. 12, no. 2, p. 32, 2024
2024
-
[45]
Optimal alignment of three sequences on a gpu,
J. Li, S. Ranka, and S. Sahni, “Optimal alignment of three sequences on a gpu,” inProceedings of the 6th International Conference on Bioinformatics and Computational Biology, 2014
2014
-
[46]
Msa-gpu: exact multiple sequence alignment using gpu,
D. Sundfeld and A. C. de Melo, “Msa-gpu: exact multiple sequence alignment using gpu,” inBrazilian Symposium on Bioinformatics. Springer, 2013, pp. 47–58
2013
-
[47]
Three- dimensional dynamic programming accelerator for multiple sequence alignment,
R.-T. Chien, Y .-L. Liao, C.-A. Wang, Y .-C. Li, and Y .-C. Lu, “Three- dimensional dynamic programming accelerator for multiple sequence alignment,” in2018 IEEE Nordic Circuits and Systems Conference (NORCAS): NORCHIP and International Symposium of System-on-Chip (SoC). IEEE, 2018, pp. 1–5
2018
-
[48]
Traceback memory reduction for three-sequence alignment algorithm with affine gap mod- els,
R.-T. Chien, M.-J. Lin, Y .-M. Yeh, and Y .-C. Lu, “Traceback memory reduction for three-sequence alignment algorithm with affine gap mod- els,” in2022 Asia-Pacific Signal and Information Processing Association Annual Summit and Conference (APSIPA ASC). IEEE, 2022, pp. 1014– 1018
2022
-
[49]
Finding functional sequence elements by multiple local alignment,
M. C. Frith, U. Hansen, J. L. Spouge, and Z. Weng, “Finding functional sequence elements by multiple local alignment,”Nucleic acids research, vol. 32, no. 1, pp. 189–200, 2004
2004
-
[50]
Ultrafast and ultralarge multiple sequence alignments using twilight,
Y .-H. Tseng, S. Walia, and Y . Turakhia, “Ultrafast and ultralarge multiple sequence alignments using twilight,”Bioinformatics, vol. 41, no. Supplement 1, pp. i332–i341, 2025
2025
-
[51]
Cuda clustalw: An efficient parallel algorithm for progressive multiple se- quence alignment on multi-gpus,
C.-L. Hung, Y .-S. Lin, C.-Y . Lin, Y .-C. Chung, and Y .-F. Chung, “Cuda clustalw: An efficient parallel algorithm for progressive multiple se- quence alignment on multi-gpus,”Computational biology and chemistry, vol. 58, pp. 62–68, 2015
2015
-
[52]
Approximate multiple protein structure align- ment using the sum-of-pairs distance,
J. Ye and R. Janardan, “Approximate multiple protein structure align- ment using the sum-of-pairs distance,”Journal of Computational Biol- ogy, vol. 11, no. 5, pp. 986–1000, 2004
2004
-
[53]
Semiglobal sequence align- ment with gaps using gpu,
T. C. Carroll, J.-T. Ojiaku, and P. W. Wong, “Semiglobal sequence align- ment with gaps using gpu,”IEEE/ACM Transactions on Computational Biology and Bioinformatics, vol. 17, no. 6, pp. 2086–2097, 2019
2086
-
[54]
Aalign: A simd framework for pairwise sequence alignment on x86-based multi-and many-core proces- sors,
K. Hou, H. Wang, and W.-c. Feng, “Aalign: A simd framework for pairwise sequence alignment on x86-based multi-and many-core proces- sors,” in2016 IEEE International Parallel and Distributed Processing Symposium (IPDPS). IEEE, 2016, pp. 780–789
2016
-
[55]
Swaphi-ls: Smith- waterman algorithm on xeon phi coprocessors for long dna sequences,
Y . Liu, T.-T. Tran, F. Lauenroth, and B. Schmidt, “Swaphi-ls: Smith- waterman algorithm on xeon phi coprocessors for long dna sequences,” in2014 IEEE International Conference on Cluster Computing (CLUS- TER). IEEE, 2014, pp. 257–265
2014
-
[56]
Graphics processing units in bioinformatics, computational biology and systems biology,
M. S. Nobile, P. Cazzaniga, A. Tangherloni, and D. Besozzi, “Graphics processing units in bioinformatics, computational biology and systems biology,”Briefings in bioinformatics, vol. 18, no. 5, pp. 870–885, 2017
2017
-
[57]
A review of biosequences alignment, matching, and mining based on gpu,
X. Kong, C. Shen, and J. Tang, “A review of biosequences alignment, matching, and mining based on gpu,”Current Bioinformatics, 2025
2025
-
[58]
Cudasw++ 3.0: accelerating smith-waterman protein database search by coupling cpu and gpu simd instructions,
Y . Liu, A. Wirawan, and B. Schmidt, “Cudasw++ 3.0: accelerating smith-waterman protein database search by coupling cpu and gpu simd instructions,”BMC bioinformatics, vol. 14, no. 1, p. 117, 2013
2013
-
[59]
Pantaleoni and N
J. Pantaleoni and N. Subtil, “Nvbio,” 2015, https://nvlabs.github.io/nvb io
2015
-
[60]
Anyseq: A high performance sequence align- ment library based on partial evaluation,
A. M ¨uller, B. Schmidt, A. Hildebrandt, R. Membarth, R. Leißa, M. Kruse, and S. Hack, “Anyseq: A high performance sequence align- ment library based on partial evaluation,” in2020 IEEE International Parallel and Distributed Processing Symposium (IPDPS). IEEE, 2020, pp. 1030–1040
2020
-
[61]
Clustalw-mpi: Clustalw analysis using distributed and parallel computing,
K.-B. Li, “Clustalw-mpi: Clustalw analysis using distributed and parallel computing,”Bioinformatics, vol. 19, no. 12, pp. 1585–1586, 2003
2003
-
[62]
Multi-gpu approach for large- scale multiple sequence alignment,
R. A. de O. Siqueira, M. A. Stefanes, L. C. Rozante, D. C. Martins- Jr, J. E. de Souza, and E. Araujo, “Multi-gpu approach for large- scale multiple sequence alignment,” inInternational Conference on Computational Science and Its Applications. Springer, 2021, pp. 560– 575
2021
-
[63]
Using reconfigurable hardware to accelerate multiple sequence alignment with clustalw,
T. Oliver, B. Schmidt, D. Nathan, R. Clemens, and D. Maskell, “Using reconfigurable hardware to accelerate multiple sequence alignment with clustalw,”Bioinformatics, vol. 21, no. 16, pp. 3431–3432, 2005
2005
-
[64]
Parallelization of the mafft multiple sequence alignment program,
K. Katoh and H. Toh, “Parallelization of the mafft multiple sequence alignment program,”Bioinformatics, vol. 26, no. 15, pp. 1899–1900, 2010
1900
-
[65]
Alisim: a fast and versatile phylogenetic sequence simulator for the genomic era,
N. Ly-Trong, S. Naser-Khdour, R. Lanfear, and B. Q. Minh, “Alisim: a fast and versatile phylogenetic sequence simulator for the genomic era,” Molecular biology and evolution, vol. 39, no. 5, p. msac092, 2022
2022
-
[66]
Fastsp: linear time calculation of alignment accuracy,
S. Mirarab and T. Warnow, “Fastsp: linear time calculation of alignment accuracy,”Bioinformatics, vol. 27, no. 23, pp. 3250–3258, 2011
2011
-
[67]
The jukes-cantor model of molecular evolution,
K. Erickson, “The jukes-cantor model of molecular evolution,”Primus, vol. 20, no. 5, pp. 438–445, 2010
2010
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.