Multi-type branching inference on contact trees with application to COVID-19
Pith reviewed 2026-06-26 18:12 UTC · model grok-4.3
The pith
Closed-form ODEs for unobserved clades and sampled-tip densities yield a likelihood for stochastic SIR on contact trees.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The authors obtain closed-form ordinary differential equations for the probability that a clade goes entirely unobserved and for the probability density that it produces an observed tip in a given state under a stochastic SIR process on a rooted contact tree; the resulting likelihood can be evaluated directly for trees with known tip states and extended to partially resolved trees by treating internal branching times as latent variables.
What carries the argument
Closed-form ODEs for the probability of an unobserved clade and the density of a sampled tip within the multi-type SIR branching process on contact trees with per-individual contact counts.
If this is right
- The likelihood evaluates exactly for any rooted contact tree whose tip states are known.
- Treating internal branching times as latent variables extends the same likelihood to partially resolved trees.
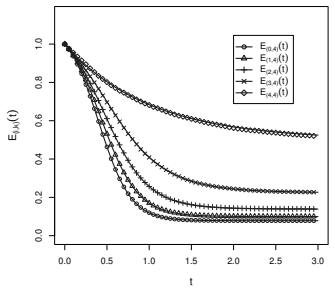
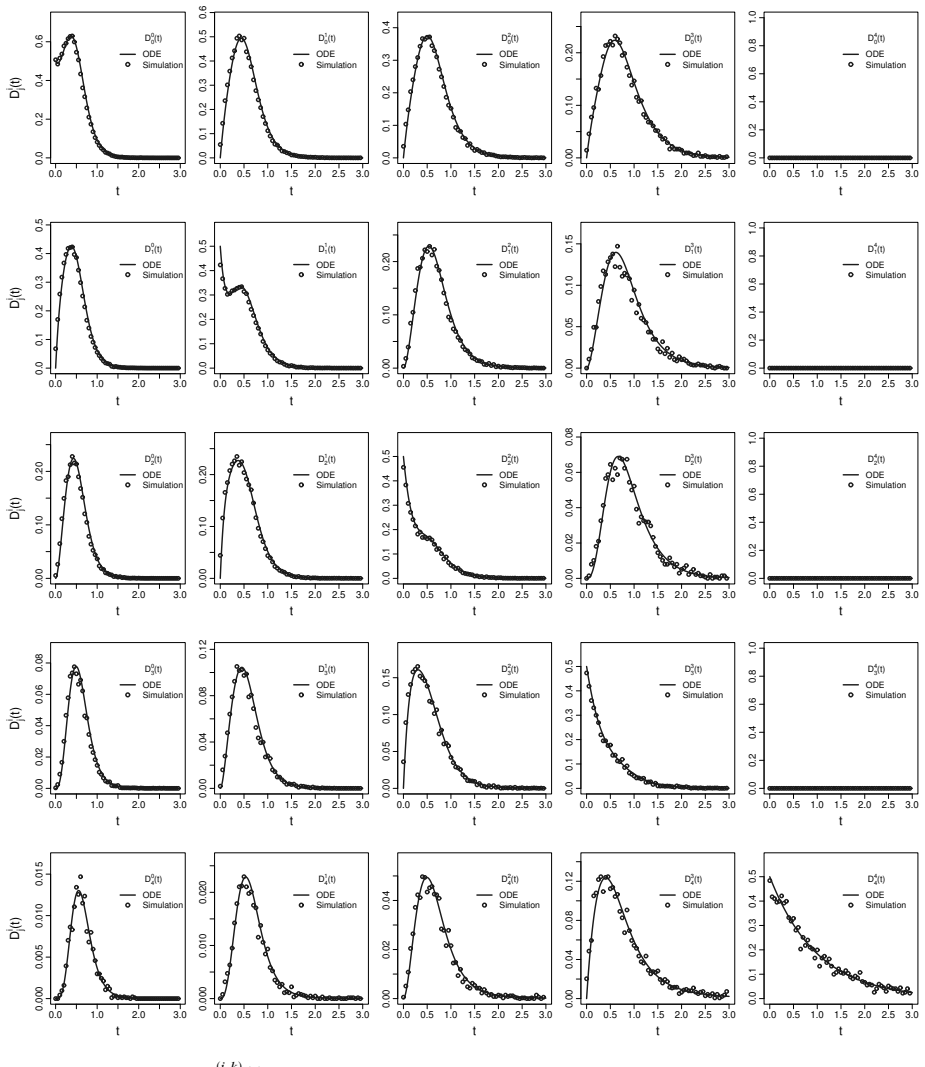
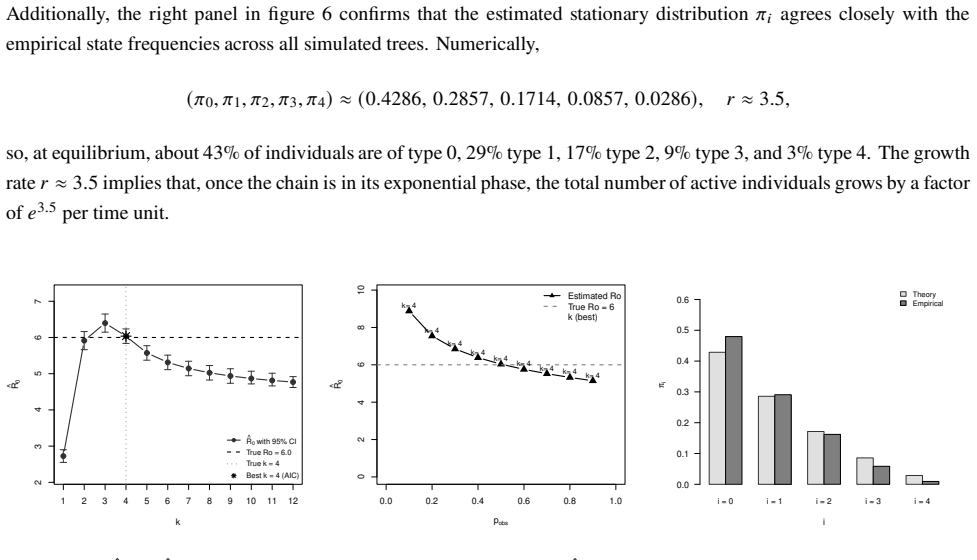
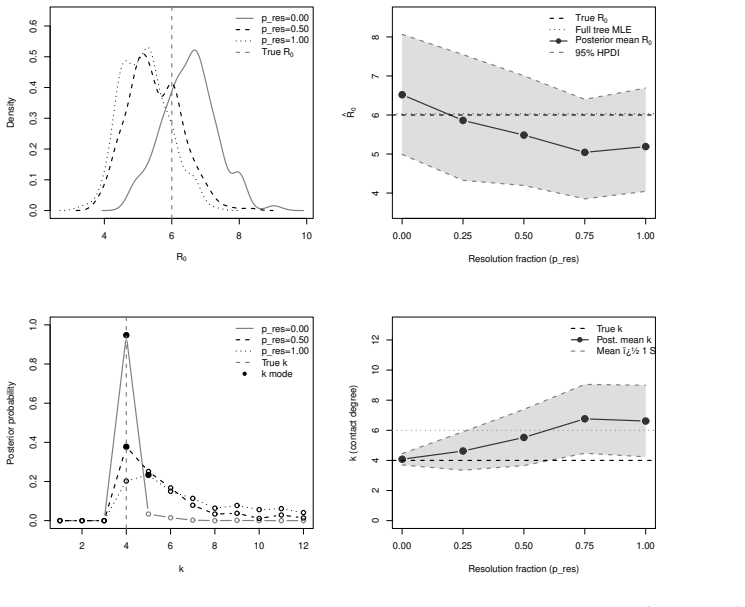
- Parameters recover accurately from simulated outbreaks with well-calibrated posterior uncertainty.
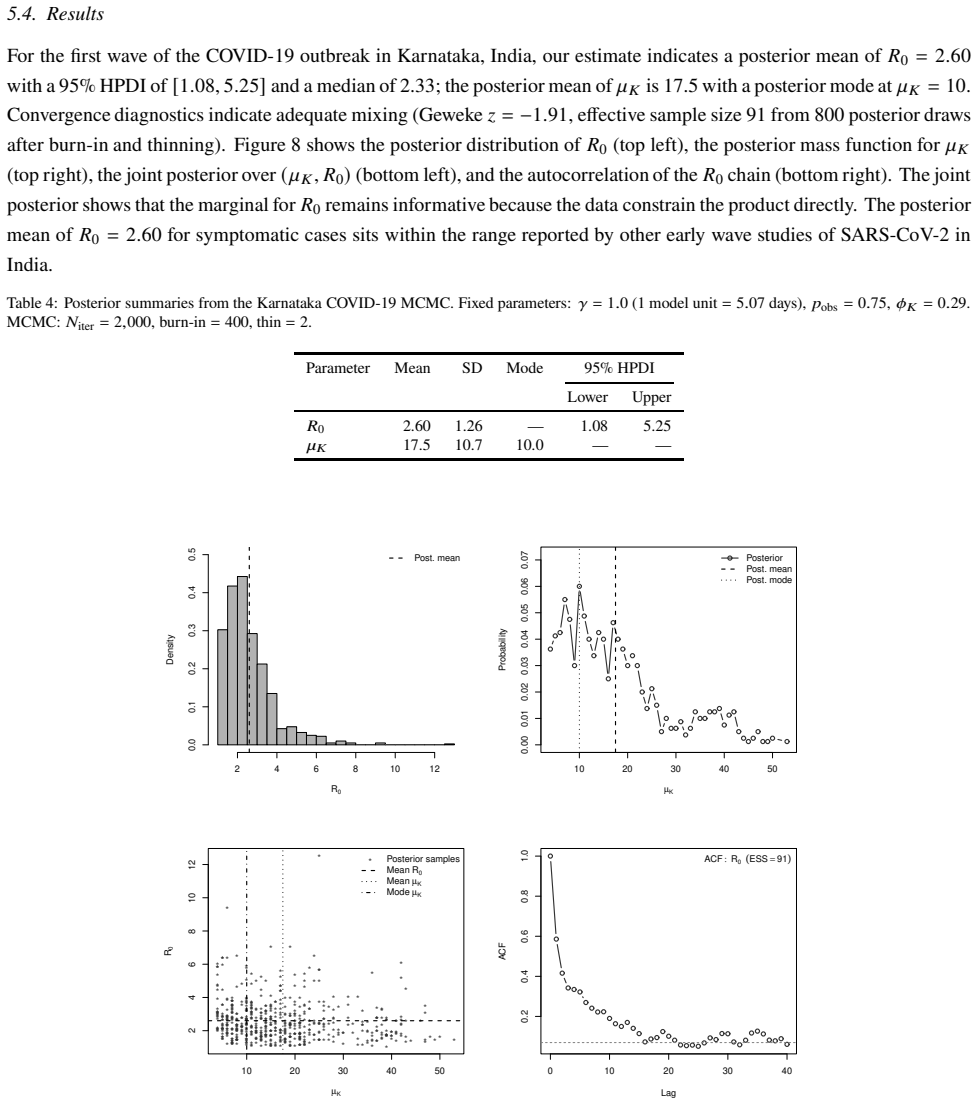
- Application to Karnataka COVID-19 contact-tracing data infers both transmission dynamics and contact-structure heterogeneity.
Where Pith is reading between the lines
- The same ODE construction could be reused for contact-tracing datasets of other directly transmitted pathogens.
- The contact-heterogeneity component could be combined with sequence-based phylodynamic models to use both tree topology and genetic data.
- Validation on real data would require checking whether reported contact trees deviate systematically from the assumed SIR branching process.
Load-bearing premise
The observed contact tree must accurately represent transmission under the stochastic SIR process in which each individual is characterized by its total effective contacts and already-infected downstream contacts.
What would settle it
Generating data under a homogeneous-mixing SIR process without individual contact counts and then fitting the derived likelihood should produce biased parameter estimates or mis-calibrated uncertainty intervals.
Figures
read the original abstract
Inferring epidemiological parameters from transmission trees is essential for understanding infectious disease dynamics. Existing tree-based likelihood methods, including the multi-type birth-death models originally applied in phylodynamic settings, provide powerful tools, but most assume homogeneous mixing and rarely capture how transmission potential changes as an individual infects more of their contacts. In this work, we develop a likelihood framework that operates directly on transmission trees, in which nodes are individuals and edges are reported transmission events, with no sequence data involved. We derive a likelihood for a stochastic SIR process on a rooted contact tree in which each infected individual is characterised by the total number of effective contacts, and the number of already infected downstream contacts. We obtain closed-form ordinary differential equations for the probability that a clade goes entirely unobserved and for the probability density that it produces an observed (sampled) tip in a given state. The resulting likelihood can be evaluated for a rooted contact tree with known tip states, and we extend it to partially resolved trees by treating internal branching times as latent variables. Validation on simulated outbreaks confirms accurate parameter recovery and well calibrated uncertainty. Application to empirical COVID-19 contact-tracing data from Karnataka, India, demonstrates the framework's utility for real epidemiological settings. By incorporating contact-degree heterogeneity in a multi-type branching likelihood, our work provides a principled baseline for inferring both transmission dynamics and contact structure from fully or partially resolved transmission trees, complementing rather than relying on sequence-based phylodynamic inference
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper develops a likelihood framework for inferring epidemiological parameters from transmission trees under a stochastic SIR process on rooted contact trees. Each infected individual is characterized by the total number of effective contacts and the number of already infected downstream contacts. Closed-form ODEs are derived for the probability that a clade goes entirely unobserved and for the probability density that it produces an observed (sampled) tip in a given state. The resulting likelihood applies to rooted contact trees with known tip states and is extended to partially resolved trees by integrating over latent internal branching times. Validation on simulated outbreaks shows accurate parameter recovery with well-calibrated uncertainty, and the method is applied to COVID-19 contact-tracing data from Karnataka, India.
Significance. If the derivations hold, the work supplies a computationally tractable multi-type branching-process likelihood that directly incorporates contact-degree heterogeneity into tree-based inference without sequence data. The closed-form ODEs for unobserved-clade and sampling probabilities constitute a technical strength, enabling efficient evaluation on observed or partially resolved trees. Simulation recovery and the Karnataka application illustrate practical utility as a complement to phylodynamic methods for contact-tracing datasets.
minor comments (3)
- [Abstract, §2] Abstract and §2: the state-space definition for the multi-type process (total contacts and downstream infections) should be stated explicitly with the resulting dimension of the ODE system to allow readers to assess computational scaling.
- [§4] §4 (simulation section): the reported parameter-recovery metrics would benefit from an explicit statement of the prior ranges used in the Bayesian inference and confirmation that the contact-tree topologies were generated under the same SIR process assumed by the likelihood.
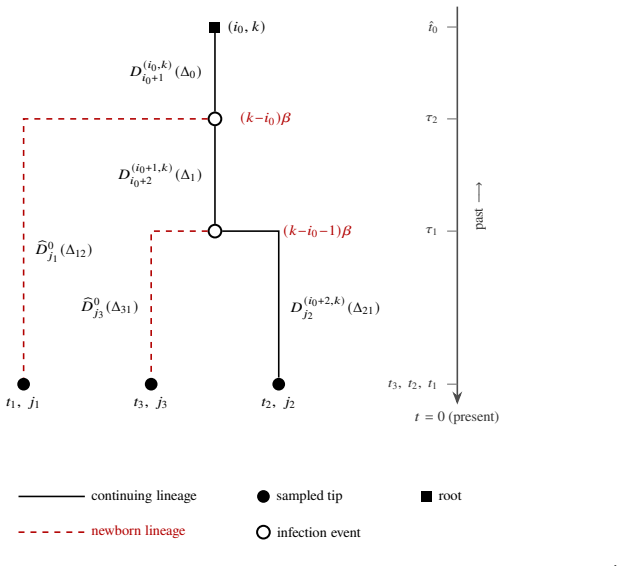
- [Figure captions, §5] Figure captions and §5 (Karnataka application): axis labels and state definitions (e.g., what constitutes an 'observed tip in a given state') should be clarified so that the plotted posterior densities can be directly compared to the model states.
Simulated Author's Rebuttal
We thank the referee for their positive summary of the manuscript, recognition of its technical contributions, and recommendation for minor revision. The provided summary accurately reflects the scope and results. No specific major comments were listed in the report.
Circularity Check
No significant circularity detected
full rationale
The central derivation consists of obtaining closed-form ODEs for the probability a clade is unobserved and the density of producing an observed tip, starting from the multi-type branching process on a contact tree with per-individual contact counts. This follows directly from the standard Kolmogorov forward equations for the process once states are defined by total effective contacts and downstream infections; no parameter is fitted to data and then relabeled as a prediction, no self-citation supplies a uniqueness theorem or ansatz, and the latent-time extension is a standard marginalization step. The framework is therefore self-contained relative to its stated model assumptions and does not reduce to its inputs by construction.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Phylodynamics on local sexual contact networks
David A Rasmussen, Roger Kouyos, Huldrych F Günthard, an d Tanja Stadler. Phylodynamics on local sexual contact networks. PLoS computational biology , 13(3):e1005448, 2017
2017
-
[2]
A multi-type branc hing process model for epidemics with application to covid-19
Arnab Kumar Laha and Sourav Majumdar. A multi-type branc hing process model for epidemics with application to covid-19. Stochastic Environmental Research and Risk Assessment, 37(1):305–325, 2023
2023
-
[3]
The impacts of spat ial–temporal heterogeneity of human-to-human contacts on the extinction probability of infectious disea se from branching process model
Wuqiong Zhao, Xia Wang, and Biao Tang. The impacts of spat ial–temporal heterogeneity of human-to-human contacts on the extinction probability of infectious disea se from branching process model. Journal of Theoretical Biology, 579:111703, 2024
2024
-
[4]
Heterogeneous network epid emics: real-time growth, variance and extinction of infection
Frank Ball and Thomas House. Heterogeneous network epid emics: real-time growth, variance and extinction of infection. Journal of Mathematical Biology , 75(3):577–619, 2017
2017
-
[5]
Multitype sir epidemics am ong a population partitioned into households with proportionate global mixing: F
Frank Ball and Liam Critcher. Multitype sir epidemics am ong a population partitioned into households with proportionate global mixing: F. ball, l. critcher. Journal of Mathematical Biology , 92(5):77, 2026
2026
-
[6]
Using multitype branching processes to quantify statistics of disease outbreaks in zoonotic epidemics
Sarabjeet Singh, David J Schneider, and Christopher R My ers. Using multitype branching processes to quantify statistics of disease outbreaks in zoonotic epidemics. Physical Review E, 89(3):032702, 2014
2014
-
[7]
Uncovering epid emiological dynamics in heterogeneous host populations using phylogenetic methods
Tanja Stadler and Sebastian Bonhoeffer. Uncovering epid emiological dynamics in heterogeneous host populations using phylogenetic methods. Philosophical Transactions of the Royal Society B: Biologi cal Sciences , 368(1614): 20120198, 2013
2013
-
[8]
Estim ating a binary character’s effect on speciation and extinction
Wayne P Maddison, Peter E Midford, and Sarah P Otto. Estim ating a binary character’s effect on speciation and extinction. Systematic biology, 56(5):701–710, 2007
2007
-
[9]
Unifying the epidemiological and evolutionary dyn amics of pathogens
Bryan T Grenfell, Oliver G Pybus, Julia R Gog, James LN Woo d, Janet M Daly, Jenny A Mumford, and Edward C Holmes. Unifying the epidemiological and evolutionary dyn amics of pathogens. science, 303(5656):327–332, 2004
2004
-
[10]
Simultaneous reconstruction of evolutionary history and epidemiological dynamics from vi ral sequences with the birth–death sir model
Denise Kühnert, Tanja Stadler, Timothy G Vaughan, and A lexei J Drummond. Simultaneous reconstruction of evolutionary history and epidemiological dynamics from vi ral sequences with the birth–death sir model. Journal of the Royal Society Interface, 11(94):20131106, 2014
2014
-
[11]
Phylody- namics of infectious disease epidemics
Erik M Volz, Sergei L Kosakovsky Pond, Melissa J Ward, An drew J Leigh Brown, and Simon DW Frost. Phylody- namics of infectious disease epidemics. Genetics, 183(4):1421–1430, 2009
2009
-
[12]
Viral phy lodynamics
Erik M Volz, Katia Koelle, and Trevor Bedford. Viral phy lodynamics. PLoS computational biology, 9(3):e1002947, 2013
2013
-
[13]
Tanja Stadler, Denise Kühnert, Sebastian Bonhoeffer, a nd Alexei J. Drummond. Birth-death skyline plot reveals temporal changes of epidemic spread in HIV and hepatitis C vi rus (HCV). Proceedings of the National Academy of Sciences, 110(1):228–233, 2013. doi: 10.1073/pnas.1207965110. 25
-
[14]
Accounting for conta ct tracing in epidemiological birth-death models
Anna Zhukova and Olivier Gascuel. Accounting for conta ct tracing in epidemiological birth-death models. PLoS Computational Biology, 21(5):e1012461, 2025
2025
-
[15]
Edge-based co mpartmental modelling for infectious disease spread
Joel C Miller, Anja C Slim, and Erik M Volz. Edge-based co mpartmental modelling for infectious disease spread. Journal of the Royal Society Interface , 9(70):890–906, 2012
2012
-
[16]
Network-based analysis o f stochastic sir epidemic models with random and proportionate mixing
Eben Kenah and James M Robins. Network-based analysis o f stochastic sir epidemic models with random and proportionate mixing. Journal of theoretical biology , 249(4):706–722, 2007
2007
-
[17]
Kiss, Joel S
István Z.. Kiss, Joel S.. Miller, and Péter Simon. Mathematics of epidemics on networks: from exact to approxi mate models. Springer International Publishing, 2017
2017
-
[18]
Phylogenies from dynamic networks
Cornelia Metzig, Oliver Ratmann, Daniela Bezemer, and Caroline Colijn. Phylogenies from dynamic networks. PLoS computational biology , 15(2):e1006761, 2019
2019
-
[19]
Exact and approx imate formulas for contact tracing on random trees
Augustine Okolie and Johannes Müller. Exact and approx imate formulas for contact tracing on random trees. Mathematical biosciences, 321:108320, 2020
2020
-
[20]
Parameter estimation for contact tracing in graph-based models
Augustine Okolie, Johannes Müller, and Mirjam Kretzschmar. Parameter estimation for contact tracing in graph-based models. Journal of the Royal Society Interface , 20(208):20230409, 2023
2023
-
[21]
Contact tracing on stochastic graphs
Augustine Okebunor Okolie. Contact tracing on stochastic graphs . PhD thesis, Technische Universität München,
- [22]
-
[23]
On incomplete sampling under birth–dea th models and connections to the sampling-based coalescent
Tanja Stadler. On incomplete sampling under birth–dea th models and connections to the sampling-based coalescent . Journal of theoretical biology , 261(1):58–66, 2009
2009
-
[24]
Contact tracing of covid-19 in karnataka, india: Superspreading and determinants of infectiousness and sym ptomatic infection
Mohak Gupta, Giridara G Parameswaran, Manraj S Sra, Ris hika Mohanta, Devarsh Patel, Amulya Gupta, Bhavik Bansal, Vardhmaan Jain, Archisman Mazumder, Mehak Arora, et al. Contact tracing of covid-19 in karnataka, india: Superspreading and determinants of infectiousness and sym ptomatic infection. Plos one, 17(7):e0270789, 2022
2022
-
[25]
Superspreading and the effect of individual variation on disease emergence
James O Lloyd-Smith, Sebastian J Schreiber, P Ekkehard Kopp, and Wayne M Getz. Superspreading and the effect of individual variation on disease emergence. Nature, 438(7066):355–359, 2005
2005
-
[26]
Kucharski, and Sebastian Funk
Akira Endo, Centre for the Mathematical Modelling of In fectious Diseases COVID-19 Working Group, Sam Abbott, Adam J. Kucharski, and Sebastian Funk. Estimating the overdispersion in covid-19 transmission using outbreak sizes outside china. Wellcome Open Research, 5:67, 2020
2020
-
[27]
Adam, Peng Wu, Jessica Y
Dillon C. Adam, Peng Wu, Jessica Y . Wong, Eric H. Y . Lau, T im K. Tsang, Simon Cauchemez, Gabriel M. Leung, and Benjamin J. Cowling. Clustering and superspreading pot ential of sars-cov-2 infections in hong kong. Nature Medicine, 26(11):1714–1719, 2020
2020
-
[28]
Modelling of reproduction number for covid-19 in india and high incide nce states
Sakthivel Marimuthu, Melvin Joy, B Malavika, Ambily Nadaraj, Edwin Sam Asirvatham, and L Jeyaseelan. Modelling of reproduction number for covid-19 in india and high incide nce states. Clinical Epidemiology and Global Health , 9:57–61, 2021
2021
-
[29]
Predictions for covid-19 outbreak in in dia using epidemiological models
Rajesh Ranjan. Predictions for covid-19 outbreak in in dia using epidemiological models. medRxiv, 2020. doi: 10.1101/2020.04.02.20051466
-
[30]
Assessment of lockdown effect in some states and overall india: A predictive mathematical study o n covid-19 outbreak
Tridip Sardar, Sk Shahid Nadim, Sourav Rana, and Joydev Chattopadhyay. Assessment of lockdown effect in some states and overall india: A predictive mathematical study o n covid-19 outbreak. Chaos, Solitons & Fractals , 139: 110078, 2020. 26
2020
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.