Affinage: genome-scale mechanistic gene annotation from the published literature
Pith reviewed 2026-07-03 01:30 UTC · model grok-4.3
The pith
Affinage uses an LLM pipeline to extract mechanistic gene functions from primary literature and annotates 19,293 human protein-coding genes.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
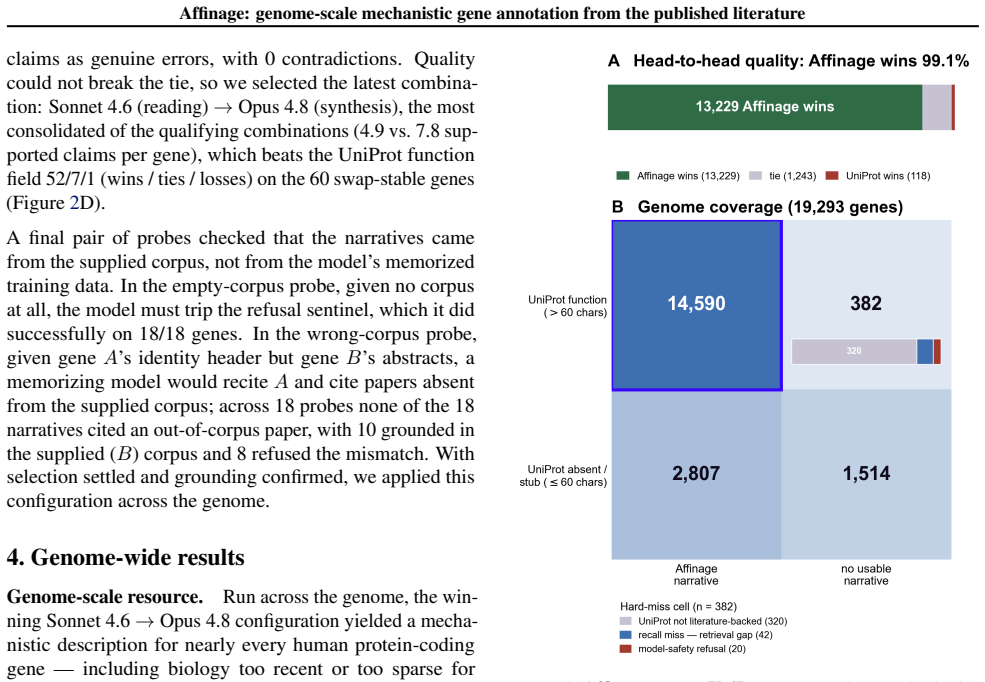
Affinage performs retrieval and mechanistic reasoning from the primary literature alone once per gene and stores the result as a reusable structured annotation. A biologist-designed reading pass extracts only direct experimental evidence and a synthesis pass reasons over those findings alone. Applied across the genome Affinage annotates 19,293 human protein-coding genes, providing mechanism for thousands whose UniProt function is empty or a stub, and beating the curated reference on 99.1 percent of head-to-head genes as scored by a cross-family LLM judge. It delineates the 10 percent of the proteome that remains mechanistically uncharacterized and releases all records openly.
What carries the argument
The Affinage pipeline, consisting of a reading pass that extracts direct experimental evidence from papers and a synthesis pass that reasons over those findings to produce structured annotations.
If this is right
- The annotations supply mechanistic details for thousands of genes where UniProt entries are empty or stub-like.
- Affinage annotations are scored higher than the curated reference on 99.1 percent of genes in head-to-head comparisons by an LLM judge.
- The pipeline identifies the 10 percent of the human proteome that remains mechanistically uncharacterized.
- All generated records are released openly as a continuously updatable literature-grounded resource.
Where Pith is reading between the lines
- The open release could let biologists directly test whether the new annotations lead to more accurate experimental hypotheses than existing database entries.
- Periodic re-running of the pipeline on updated literature could produce a living census that tracks how gene function knowledge evolves over time.
- The separation of reading and synthesis passes might be adapted to annotate other biological entities such as pathways or protein complexes.
Load-bearing premise
That an LLM-based judge can reliably and without bias determine which annotation supplies better mechanistic insight when comparing Affinage outputs to existing database entries.
What would settle it
Direct evaluation by human biologists of the mechanistic accuracy and experimental grounding of Affinage annotations versus UniProt entries for a sample of genes previously lacking detailed function descriptions.
Figures
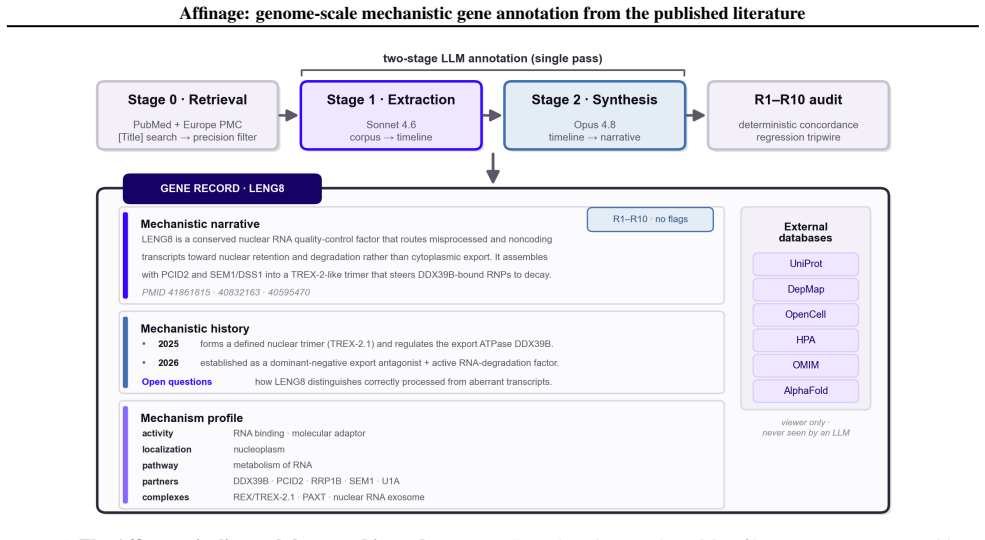
read the original abstract
Understanding the mechanistic function of a gene is a critical starting point for biology. However, for much of the human proteome that knowledge is scattered across thousands of primary papers or remains poorly established, while the curated databases biologists rely on can lag years behind recent literature. Large language models can now read and synthesize that literature on demand, but doing so faithfully for many genes is an expensive, non-reproducible retrieval session that does not scale across users. Here, we present Affinage, an LLM pipeline that performs this retrieval and mechanistic reasoning once per gene--from the primary literature alone--and stores the result as a reusable, structured annotation. A biologist-designed reading pass extracts only direct experimental evidence, and a synthesis pass reasons over those findings alone. Applied across the genome, Affinage annotates 19,293 human protein-coding genes. This analysis provides mechanism for thousands of genes whose UniProt function is empty or a stub, beating the curated reference on 99.1% of head-to-head genes as scored by a cross-family LLM judge. Affinage also delineates the 10% of the proteome that remains mechanistically uncharacterized and will serve as a continuously-updated, literature-grounded census of gene function. All records are released openly at https://affinage.wi.mit.edu . More broadly, Affinage serves as an example of how domain experts can encode their expertise into scalable LLM pipelines to improve the publicly available data that guides biological hypotheses and experimentation.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents Affinage, an LLM pipeline that extracts mechanistic gene annotations directly from primary literature via a biologist-designed reading pass (limited to experimental evidence) followed by a synthesis pass. Applied to the human proteome, it produces structured annotations for 19,293 protein-coding genes, supplies mechanistic detail for thousands of genes with empty or stub UniProt entries, and reports that these annotations are superior to UniProt on 99.1% of head-to-head genes according to a cross-family LLM judge. All records are released openly.
Significance. If the evaluation methodology is shown to be reliable, the work provides a scalable, literature-grounded approach to updating gene-function annotations at genome scale, addressing the lag between primary literature and curated databases. The open release of the full set of annotations is a clear strength that would enable community use and further validation.
major comments (2)
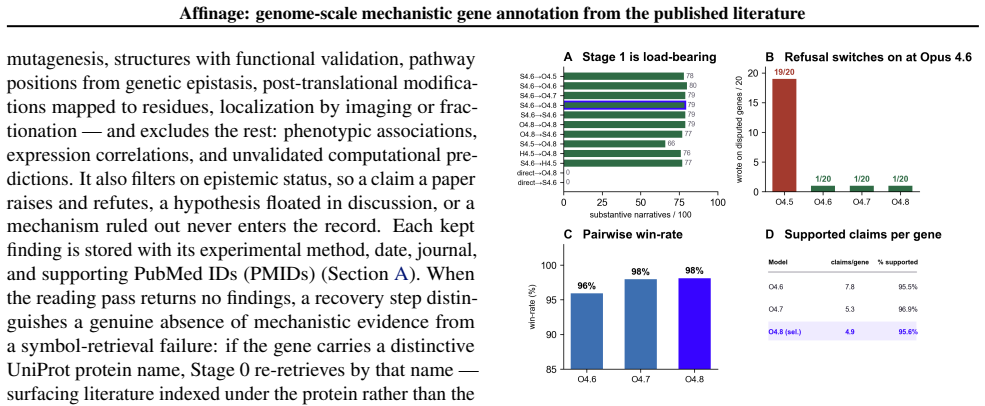
- [Abstract / Results] Abstract and Results (performance comparison): The central claim that Affinage outperforms UniProt on 99.1% of head-to-head genes rests entirely on scores from a cross-family LLM judge. No section describes calibration of this judge against human experts, inter-rater agreement rates, or explicit tests for systematic bias favoring LLM-generated mechanistic phrasing over curated entries. Because both the annotations and the evaluator are LLM-based, this unverified component is load-bearing for the superiority result.
- [Methods] Methods (LLM extraction and synthesis passes): The manuscript states that the reading pass extracts only direct experimental evidence and the synthesis pass reasons over those findings alone, yet provides no quantitative validation (e.g., precision/recall against a human-annotated gold set) of hallucination rates or fidelity to source papers for either pass. This directly affects the trustworthiness of the 19,293 annotations released as the primary output.
minor comments (2)
- [Methods] The manuscript would benefit from an explicit statement of the exact LLM models, temperature settings, and prompt templates used in both the annotation pipeline and the judge, ideally with version numbers for reproducibility.
- [Results] Figure or table presenting the head-to-head comparison should include the total number of genes evaluated, the breakdown by UniProt annotation quality (empty vs. stub), and confidence intervals on the 99.1% figure.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback emphasizing the need for rigorous validation of the LLM components. We address each major comment below and have revised the manuscript to improve transparency on evaluation methodology while acknowledging limitations.
read point-by-point responses
-
Referee: [Abstract / Results] Abstract and Results (performance comparison): The central claim that Affinage outperforms UniProt on 99.1% of head-to-head genes rests entirely on scores from a cross-family LLM judge. No section describes calibration of this judge against human experts, inter-rater agreement rates, or explicit tests for systematic bias favoring LLM-generated mechanistic phrasing over curated entries. Because both the annotations and the evaluator are LLM-based, this unverified component is load-bearing for the superiority result.
Authors: We agree that the superiority claim depends on the LLM judge without reported human calibration, inter-rater agreement, or explicit bias tests in the original manuscript. The cross-family judge was selected specifically to reduce stylistic favoritism toward LLM output. In revision we will expand the Methods to fully document the judge prompt, add a dedicated subsection on potential biases and the rationale for this evaluator, and include results from a new small-scale human calibration study on a random sample of 50 genes. This directly addresses the load-bearing nature of the evaluation. revision: yes
-
Referee: [Methods] Methods (LLM extraction and synthesis passes): The manuscript states that the reading pass extracts only direct experimental evidence and the synthesis pass reasons over those findings alone, yet provides no quantitative validation (e.g., precision/recall against a human-annotated gold set) of hallucination rates or fidelity to source papers for either pass. This directly affects the trustworthiness of the 19,293 annotations released as the primary output.
Authors: The manuscript does not report quantitative precision/recall or hallucination rates against a human gold standard. This is a genuine gap. The reading and synthesis passes were engineered with biologist-specified constraints to limit outputs to direct experimental evidence, but without a gold set we cannot quantify fidelity at scale. In revision we will add an explicit Limitations section discussing this absence, the prompt-based safeguards employed, and the decision to release all source-paper links to support independent verification. A full genome-scale human gold set is not feasible within current resources, but the open release enables community validation. revision: partial
- Provision of a comprehensive human-annotated gold set and associated precision/recall metrics for hallucination and fidelity across all 19,293 genes
Circularity Check
No circularity: evaluation step is independent of annotation pipeline
full rationale
The paper describes an LLM pipeline that extracts experimental evidence from literature and synthesizes mechanistic annotations for genes. The 99.1% superiority claim is presented as an outcome of applying a separate cross-family LLM judge to compare outputs against UniProt entries. No equations, fitted parameters, or self-citations are quoted that reduce the annotation result or the superiority metric to the inputs by construction. The judge is described as an external scoring mechanism rather than a self-definitional loop or renamed fit. The derivation chain for producing annotations remains self-contained against external literature sources and does not collapse into its own evaluation step.
Axiom & Free-Parameter Ledger
free parameters (1)
- LLM model choice and prompt design
axioms (1)
- domain assumption Large language models can be prompted to extract only direct experimental evidence without introducing unsupported inferences.
Reference graph
Works this paper leans on
-
[1]
A., Balhoff, J., Carbon, S., Cherry, J
Aleksander, S. A., Balhoff, J., Carbon, S., Cherry, J. M., Drabkin, H. J., Ebert, D., Feuermann, M., Gaudet, P., Harris, N. L., et al. The G ene O ntology knowledgebase in 2023. Genetics, 224 0 (1): 0 iyad031, 2023. doi:10.1093/genetics/iyad031
-
[2]
Claude S onnet 4.6 and O pus 4.6 model card
Anthropic . Claude S onnet 4.6 and O pus 4.6 model card. Technical report, https://www.anthropic.com/, 2025
2025
-
[3]
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., Davis, A. P., Dolinski, K., Dwight, S. S., Eppig, J. T., et al. Gene ontology: tool for the unification of biology. Nature Genetics, 25 0 (1): 0 25--29, 2000. doi:10.1038/75556
-
[4]
Chen, Y. and Zou, J. GenePert : Leveraging GenePT embeddings for gene perturbation prediction. bioRxiv, 2024. doi:10.1101/2024.10.27.620513
-
[5]
Chen, Y. T. and Zou, J. Simple and effective embedding model for single-cell biology built from ChatGPT . Nature Biomedical Engineering, 9 0 (4): 0 483--493, 2025. doi:10.1038/s41551-024-01293-z
-
[6]
Europe PMC : a full-text literature database for the life sciences and platform for innovation
Europe PMC Consortium . Europe PMC : a full-text literature database for the life sciences and platform for innovation. Nucleic Acids Research, 43 0 (D1): 0 D1042--D1048, 2015. doi:10.1093/nar/gku1061
-
[7]
Biomni : A general-purpose biomedical AI agent
Huang, K., Zhang, S., Wang, H., Qu, Y., et al. Biomni : A general-purpose biomedical AI agent. bioRxiv, 2025. doi:10.1101/2025.05.30.656746
-
[8]
Hutchins, B. I., Yuan, X., Anderson, J. M., and Santangelo, G. M. Relative C itation R atio ( RCR ): A new metric that uses citation rates to measure influence at the article level. PLOS Biology, 14 0 (9): 0 e1002541, 2016. doi:10.1371/journal.pbio.1002541
-
[9]
A large language model framework for literature-based disease--gene association prediction
Li, P.-H., Sun, Y.-Y., Juan, H.-F., Chen, C.-Y., Tsai, H.-K., and Huang, J.-H. A large language model framework for literature-based disease--gene association prediction. Briefings in Bioinformatics, 26 0 (1): 0 bbaf070, 2025. doi:10.1093/bib/bbaf070
-
[10]
Mitchener, L. et al. Kosmos : An AI scientist for autonomous discovery. arXiv preprint arXiv:2511.02824, 2025. doi:10.48550/arXiv.2511.02824
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv.2511.02824 2025
-
[11]
Sayers, E. W., Bolton, E. E., Brister, J. R., Canese, K., Chan, J., Comeau, D. C., Connor, R., Funk, K., Kelly, C., Kim, S., et al. Database resources of the N ational C enter for B iotechnology I nformation. Nucleic Acids Research, 50 0 (D1): 0 D20--D26, 2022. doi:10.1093/nar/gkab1112
-
[12]
L., Braschi, B., Gray, K., Jones, T
Seal, R. L., Braschi, B., Gray, K., Jones, T. E. M., Tweedie, S., Haim-Vilmovsky, L., and Bruford, E. A. Genenames.org: the HGNC resources in 2023. Nucleic Acids Research, 51 0 (D1): 0 D1003--D1009, 2023. doi:10.1093/nar/gkac1062
-
[13]
UniProt : the U niversal P rotein K nowledgebase in 2023
The UniProt Consortium . UniProt : the U niversal P rotein K nowledgebase in 2023. Nucleic Acids Research, 51 0 (D1): 0 D523--D531, 2023. doi:10.1093/nar/gkac1052
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.