Multiscale reconstruction of protein conformations from cryo-EM images
Pith reviewed 2026-06-26 22:17 UTC · model grok-4.3
The pith
A multiscale algorithm using explicit protein backbone representation recovers atomic structures from noisy cryo-EM images.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
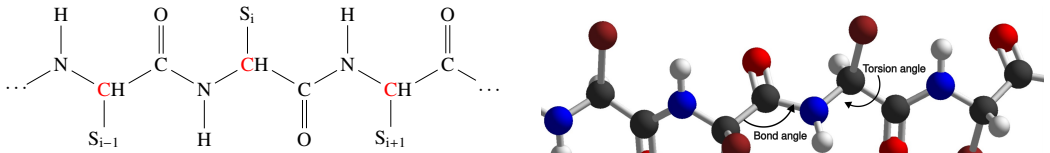
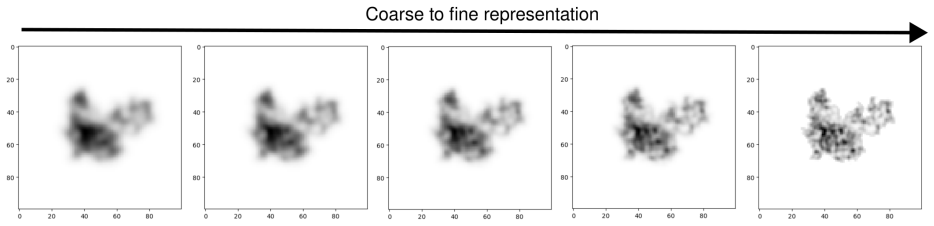
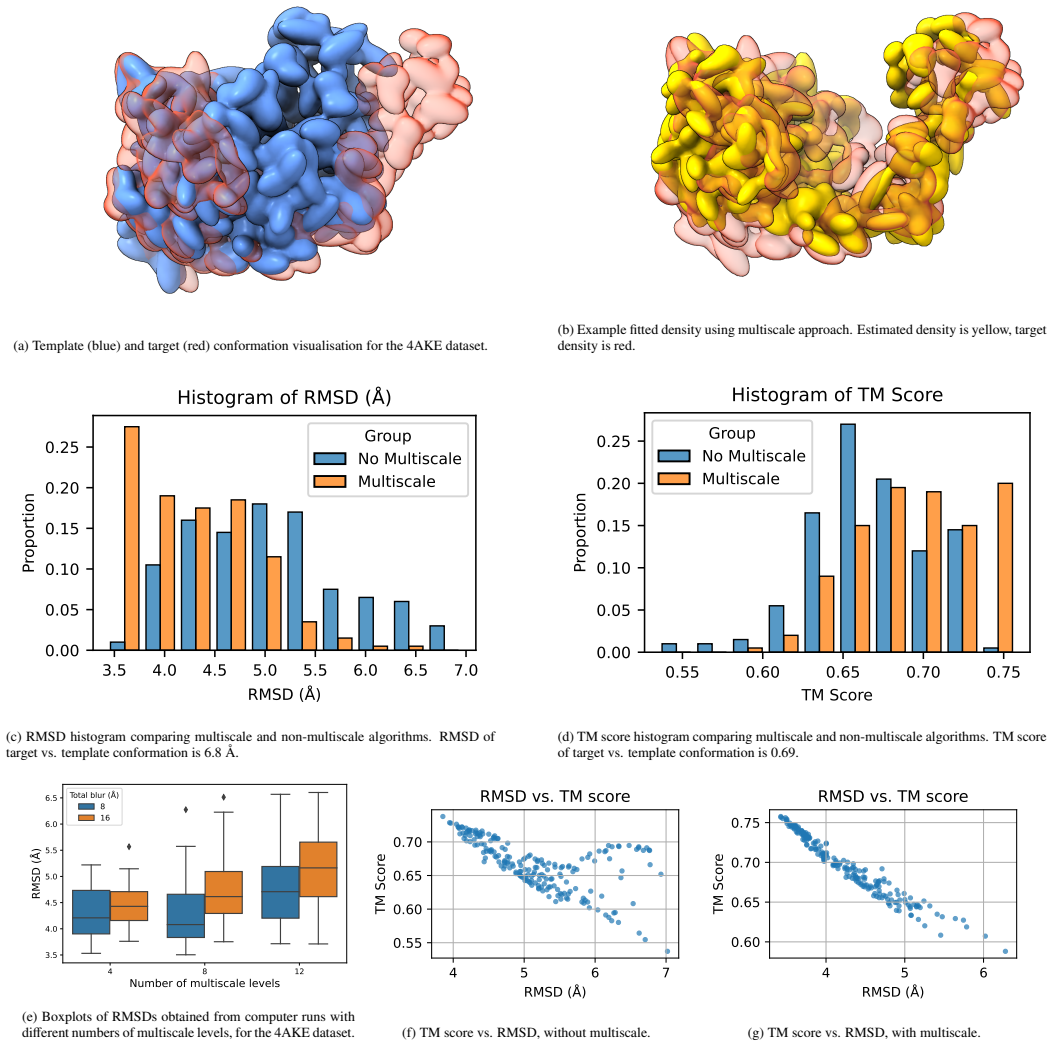
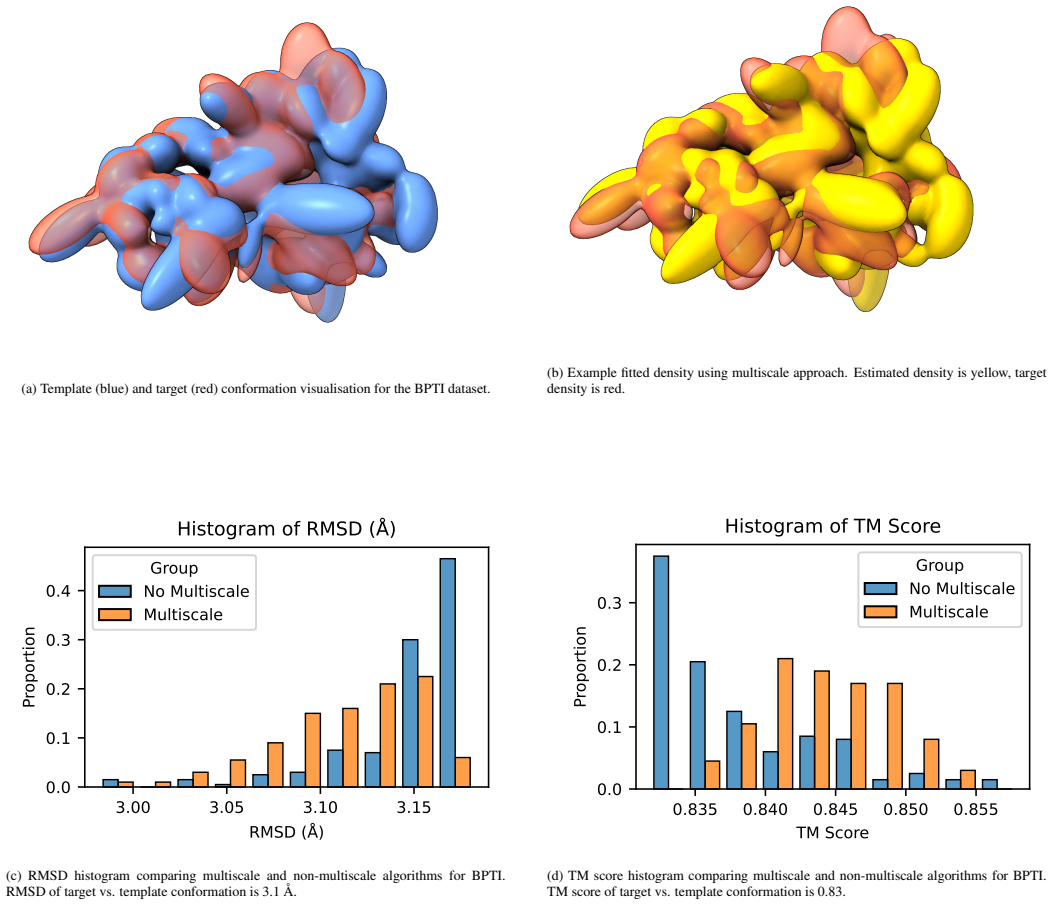
We present a novel multiscale algorithm for directly recovering the atomic model structure of a protein from single-particle cryo-EM data. Our algorithm is able to estimate protein structures to state-of-the-art accuracy for high-noise and low-contrast data. It is also robust to misspecifications in the TEM image formation model. These desirable properties are primarily due to the use of an explicit representation of the protein backbone in terms of bonds, torsion angles and bond angles, which supplies rich prior information to the structure recovery process. We apply our method on three protein cryo-EM datasets, generated using an electron microscope digital twin, and show that using a mult
What carries the argument
multiscale optimization guided by an explicit protein backbone model expressed through bonds, torsion angles, and bond angles
If this is right
- The algorithm achieves state-of-the-art accuracy on high-noise and low-contrast cryo-EM data.
- Performance remains stable even when the TEM image formation model is misspecified.
- Multiscale processing improves RMSD and TM scores relative to non-multiscale baselines on the tested datasets.
- Larger-scale structures are recovered first, lowering the chance of convergence to bad local minima.
- The method works directly on single-particle cryo-EM data to produce atomic models.
Where Pith is reading between the lines
- The backbone prior could be combined with other geometric constraints to handle even lower signal-to-noise ratios.
- Similar multiscale strategies might transfer to related inverse problems such as tomography of non-protein macromolecules.
- If the backbone representation proves sufficient on real experimental images, it would reduce reliance on high-resolution reference maps during initial modeling.
Load-bearing premise
The explicit representation of the protein backbone in terms of bonds, torsion angles and bond angles supplies rich prior information to the structure recovery process that is sufficient to guide multiscale optimization away from bad local minima.
What would settle it
Running the same three simulated datasets through the algorithm both with and without the multiscale schedule and comparing the resulting RMSD and TM scores to ground truth would show whether the reported gains are due to the multiscale strategy.
Figures


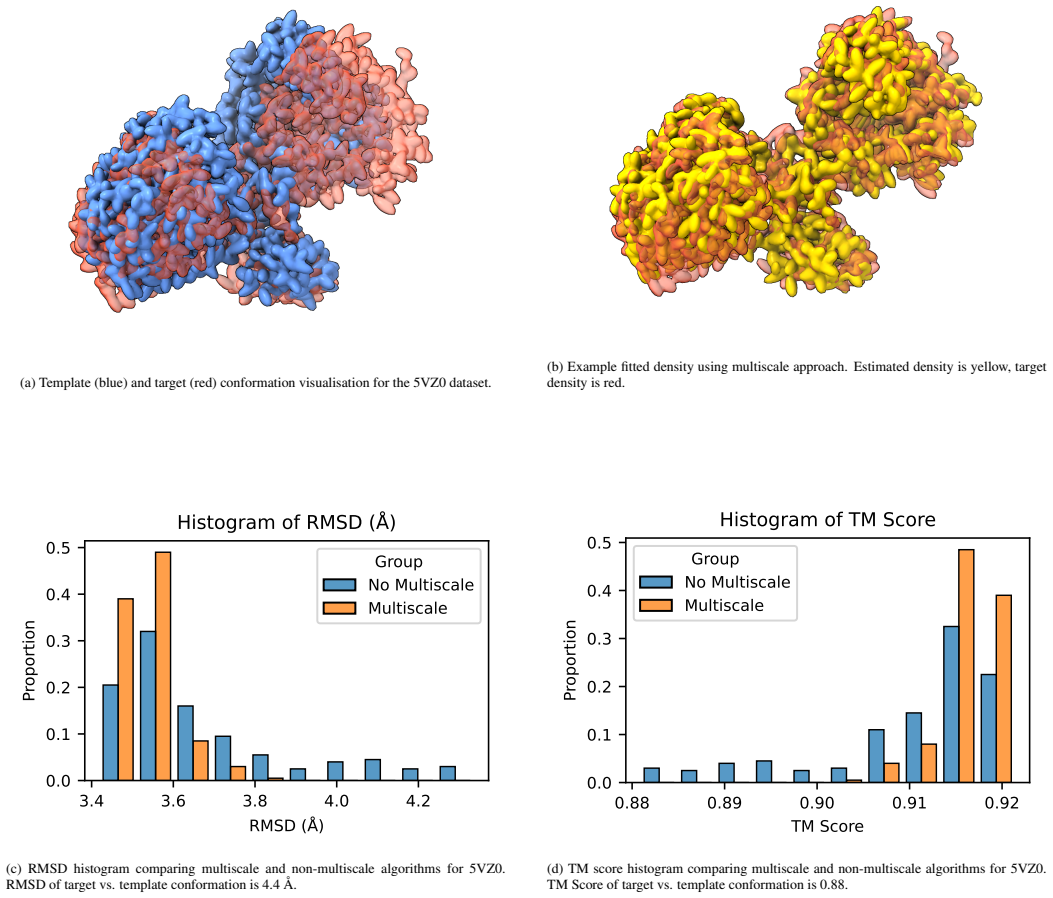
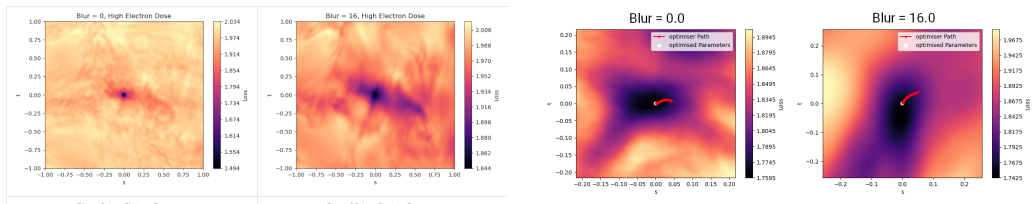

read the original abstract
We present a novel multiscale algorithm for directly recovering the atomic model structure of a protein from single-particle cryo-EM data. Our algorithm is able to estimate protein structures to state-of-the-art accuracy for high-noise and low-contrast data. It is also robust to misspecifications in the TEM image formation model. These desirable properties are primarily due to the use of an explicit representation of the protein backbone in terms of bonds, torsion angles and bond angles, which supplies rich prior information to the structure recovery process. We apply our method on three protein cryo-EM datasets, generated using an electron microscope digital twin, and show that using a multiscale approach yields an improvement of the root-mean-square deviation (RMSD) and template modelling (TM) scores with respect to the ground truth. Furthermore, there is evidence that larger-scale structures are being prioritised with the multiscale algorithm, which reduces the possibility of convergence to bad local minima.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper presents a multiscale algorithm for directly recovering atomic protein structures from single-particle cryo-EM data. It uses an explicit backbone representation parameterized by bonds, torsion angles, and bond angles to supply structural priors, claims state-of-the-art accuracy on high-noise/low-contrast data and robustness to TEM image-formation misspecifications, and reports RMSD/TM-score improvements on three simulated datasets generated by an electron-microscope digital twin, attributing gains to the multiscale strategy and backbone model.
Significance. If validated beyond the simulator, the explicit geometric backbone prior combined with multiscale optimization could provide a useful route to better convergence in low-SNR cryo-EM reconstruction. The approach is distinguished by its direct use of bond/torsion constraints rather than implicit density-based methods, but the current evidence base is confined to internal digital-twin experiments.
major comments (3)
- [Abstract / Experiments] Abstract and Experiments section: the reported RMSD and TM-score improvements are stated without numerical values, error bars, baseline comparisons, or ablation studies, preventing assessment of whether the multiscale backbone method actually reaches the claimed state-of-the-art accuracy.
- [Abstract / Experiments] Abstract and data-generation description: all robustness claims to TEM-model misspecifications rest on tests performed inside the same digital twin used to synthesize the three datasets; this leaves real-world effects (detector response, beam-induced motion, unknown aberrations, conformational heterogeneity outside the backbone parametrization) unexamined and makes the generalization claim load-bearing but unsupported.
- [Abstract] Abstract: the central attribution of performance gains to the explicit backbone prior and multiscale optimization is supported only by the three simulated cases; no additional controls, real-data tests, or analysis of local-minima avoidance are provided to substantiate that the prior is sufficient to steer optimization away from bad minima on experimental micrographs.
Simulated Author's Rebuttal
We thank the referee for the detailed and constructive report. We address each major comment below, indicating planned revisions where appropriate.
read point-by-point responses
-
Referee: [Abstract / Experiments] Abstract and Experiments section: the reported RMSD and TM-score improvements are stated without numerical values, error bars, baseline comparisons, or ablation studies, preventing assessment of whether the multiscale backbone method actually reaches the claimed state-of-the-art accuracy.
Authors: We agree that explicit numerical values, error bars, baseline comparisons, and ablation studies are needed for proper assessment. The experiments section reports the improvements but we will revise both the abstract and experiments to include the specific RMSD and TM-score values with statistics, direct baseline comparisons, and ablation results demonstrating the multiscale contribution. revision: yes
-
Referee: [Abstract / Experiments] Abstract and data-generation description: all robustness claims to TEM-model misspecifications rest on tests performed inside the same digital twin used to synthesize the three datasets; this leaves real-world effects (detector response, beam-induced motion, unknown aberrations, conformational heterogeneity outside the backbone parametrization) unexamined and makes the generalization claim load-bearing but unsupported.
Authors: The robustness tests were performed inside the digital twin to enable controlled evaluation of image-formation misspecifications. We acknowledge that this does not address all real-world effects such as beam-induced motion or conformational heterogeneity. In revision we will temper the generalization language in the abstract and add an explicit limitations discussion on the simulator's scope. revision: partial
-
Referee: [Abstract] Abstract: the central attribution of performance gains to the explicit backbone prior and multiscale optimization is supported only by the three simulated cases; no additional controls, real-data tests, or analysis of local-minima avoidance are provided to substantiate that the prior is sufficient to steer optimization away from bad minima on experimental micrographs.
Authors: The attribution rests on the three simulated datasets and the observed prioritization of larger-scale structures. We will add analysis of optimization trajectories to illustrate local-minima behavior in the revision. Real-data tests with unknown ground truth lie outside the current scope, which focuses on quantitative evaluation against known atomic models. revision: partial
- Validation on real (non-simulated) experimental cryo-EM data
Circularity Check
No circularity in derivation chain
full rationale
The paper describes a multiscale optimization algorithm that uses an explicit backbone parametrization (bonds, torsion angles, bond angles) as prior information to recover protein structures from cryo-EM images. No equations, parameter-fitting steps, or self-citations are presented in the provided text that would reduce the reported RMSD/TM improvements or robustness claims to quantities defined by the same fitted parameters or by construction. The central claims rest on empirical results from simulated data rather than any self-referential derivation, making the algorithm's logic self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Protein backbone geometry (fixed bonds, variable torsion and bond angles) supplies sufficient prior information to improve reconstruction from noisy cryo-EM data.
Reference graph
Works this paper leans on
-
[1]
Proceedings of the 33rd
Paszke, Adam and Gross, Sam and Massa, Francisco and Lerer, Adam and Bradbury, James and Chanan, Gregory and Killeen, Trevor and Lin, Zeming and Gimelshein, Natalia and Antiga, Luca and Desmaison, Alban and Köpf, Andreas and Yang, Edward and DeVito, Zach and Raison, Martin and Tejani, Alykhan and Chilamkurthy, Sasank and Steiner, Benoit and Fang, Lu and B...
-
[2]
, month = jul, year =
Zhong, Ellen and Lerer, Adam and Davis, Joey and Berger, B. , month = jul, year =. Exploring generative atomic models in cryo-
-
[3]
Jamali, Kiarash and Kimanius, Dari and Scheres, Sjors HW , month = feb, year =. A
-
[4]
Gupta, Harshit and Phan, Thong Huy and Yoo, Jaejun and Unser, Michael , month = jul, year =. Multi-. Computer
-
[6]
Advances in neural information processing systems , author =
Amortized. Advances in neural information processing systems , author =. 2022 , pmid =
2022
-
[7]
Nature Methods , author =. 2021 , note =. doi:10.1038/s41592-020-01049-4 , abstract =
-
[8]
Zhong, Ellen and Lerer, Adam and Davis, Joseph H. and Berger, Bonnie , month = oct, year =. 2021. doi:10.1109/ICCV48922.2021.00403 , abstract =
-
[9]
arXiv preprint , author =
Inferring a. arXiv preprint , author =
-
[10]
Cryo-electron microscopy – a primer for the non-microscopist , volume =. The FEBS Journal , author =. 2013 , keywords =. doi:10.1111/febs.12078 , abstract =
-
[11]
Macromolecular structure determination using. Acta Crystallographica. Section D, Structural Biology , author =. 2019 , pmid =. doi:10.1107/S2059798319011471 , abstract =
-
[12]
IEEE Transactions on Computational Imaging , author =. 2021 , keywords =. doi:10.1109/TCI.2021.3096491 , abstract =
-
[13]
International Conference on Information Fusion , author =
A look at. International Conference on Information Fusion , author =. 2011 , pages =
2011
-
[14]
Beckstein, Oliver and Seyler, Sean L. and. Simulated trajectory ensembles for the closed-to-open transition of adenylate kinase from. 2018 , keywords =. doi:10.6084/M9.FIGSHARE.7165306.V2 , abstract =
-
[15]
Acta Crystallographica Section D: Structural Biology , author =
Real-space refinement in. Acta Crystallographica Section D: Structural Biology , author =. 2018 , pages =. doi:10.1107/S2059798318006551 , abstract =
-
[16]
Acta Crystallographica Section D Biological Crystallography , author =. 2010 , pages =. doi:10.1107/S0907444909052925 , abstract =
-
[17]
bioRxiv preprint , publisher =
Ducrocq, Gabriel and Grunewald, Lukas and Westenhoff, Sebastian and Lindsten, Fredrik , month = jun, year =. bioRxiv preprint , publisher =. doi:10.1101/2024.06.19.599686 , abstract =
-
[18]
Use of multivariate statistics in analysing the images of biological macromolecules , volume =. Ultramicroscopy , author =. 1981 , pages =. doi:10.1016/0304-3991(81)90059-0 , language =
-
[19]
arXiv preprint , author =
Protein. arXiv preprint , author =
-
[20]
SIAM Journal on Imaging Sciences , author =
Structural. SIAM Journal on Imaging Sciences , author =. 2018 , pages =. doi:10.1137/17M1153509 , language =
-
[21]
Cryo-. Inverse Problems , author =. 2020 , pages =. doi:10.1088/1361-6420/ab4f55 , abstract =
-
[22]
Spectral decomposition of atomic structures in heterogeneous cryo-. Inverse Problems , author =. 2023 , pages =. doi:10.1088/1361-6420/acb2ba , abstract =
-
[23]
SIAM Journal on Imaging Sciences , author =
Regularizing. SIAM Journal on Imaging Sciences , author =. 2023 , pages =. doi:10.1137/22M1520773 , language =
-
[24]
Manuscript in preparation , author =
Multiscale reconstruction of protein conformations from cryo-. Manuscript in preparation , author =
-
[25]
Dashti, A. and Shekhar, M. S. and Hail, D. Ben and Mashayekhi, G. and Schwander, P. and Georges, A. des and Frank, J. and Singharoy, A. and Ourmazd, A. , month = jan, year =. Functional. bioRxiv preprint , publisher =. doi:10.1101/291922 , abstract =
-
[26]
Nature Methods , author =. 2024 , pages =. doi:10.1038/s41592-024-02486-1 , language =
-
[27]
Sigworth, Fred , year =
-
[28]
Kirkland, E. J. , year =. Advanced computing in electron microscopy , publisher =
-
[29]
I-. Nature Protocols , author =. 2010 , pages =. doi:10.1038/nprot.2010.5 , language =
-
[30]
Proteins: Structure, Function, and Bioinformatics , author =
Scoring function for automated assessment of protein structure template quality , volume =. Proteins: Structure, Function, and Bioinformatics , author =. 2004 , pages =. doi:10.1002/prot.20264 , abstract =
-
[31]
Acta Crystallographica Section D Biological Crystallography , author =
The. Acta Crystallographica Section D Biological Crystallography , author =. 2006 , pages =. doi:10.1107/S0907444906022116 , number =
-
[32]
Acta Crystallographica Section D Biological Crystallography , author =
Features and development of. Acta Crystallographica Section D Biological Crystallography , author =. 2010 , pages =. doi:10.1107/S0907444910007493 , abstract =
-
[33]
Protein Science , author =. 2023 , pages =. doi:10.1002/pro.4792 , abstract =
-
[34]
Nucleic Acids Research , author =. 2024 , pages =. doi:10.1093/nar/gkad1084 , abstract =
-
[35]
Lindeberg, Tony , year =. Scale-. Wiley. doi:10.1002/9780470050118.ecse609 , note =
-
[36]
and Burt, P
Adelson, E. and Burt, P. and Anderson, C. and Ogden, J. M. and Bergen, J. , month = nov, year =
-
[37]
Kirkland, Earl J. , year =. Advanced. doi:10.1007/978-1-4419-6533-2 , keywords =
-
[38]
Fragmentation. Chemical Reviews , author =. 2012 , note =. doi:10.1021/cr200093j , number =
-
[39]
Discrete. Physical Review E , author =. 2011 , note =. doi:10.1103/PhysRevE.83.061908 , abstract =
-
[40]
Methods in Molecular Biology (Clifton, N.J.) , author =
Methods of protein structure comparison , volume =. Methods in Molecular Biology (Clifton, N.J.) , author =. 2012 , pmid =. doi:10.1007/978-1-61779-588-6_10 , abstract =
-
[41]
PLOS Computational Biology , author =
Path. PLOS Computational Biology , author =. 2015 , note =. doi:10.1371/journal.pcbi.1004568 , abstract =
-
[42]
Ultramicroscopy , author =. 2015 , keywords =. doi:10.1016/j.ultramic.2015.04.016 , abstract =
-
[43]
Parakeet: a digital twin software pipeline to assess the impact of experimental parameters on tomographic reconstructions for cryo-electron tomography , volume =. Open Biology , author =. 2021 , note =. doi:10.1098/rsob.210160 , abstract =
-
[44]
Nature Methods , author =. 2024 , note =. doi:10.1038/s41592-023-02099-0 , abstract =
-
[45]
IEEE Journal of Selected Topics in Signal Processing , author =
Coarse-. IEEE Journal of Selected Topics in Signal Processing , author =. 2016 , note =. doi:10.1109/JSTSP.2015.2489186 , abstract =
-
[46]
Information and Inference: A Journal of the IMA , author =
Sketching for large-scale learning of mixture models , volume =. Information and Inference: A Journal of the IMA , author =. 2018 , pages =. doi:10.1093/imaiai/iax015 , abstract =
-
[47]
Gaussian mixture reduction via clustering , author =
-
[48]
Assa, A. and Plataniotis, K. , year =. Wasserstein-. doi:10.1109/LSP.2018.2865829 , journal =
-
[49]
Runnalls, A. R. , year =. Kullback-. doi:10.1109/TAES.2007.4383588 , journal =
-
[50]
Zhang, Qiong and Zhang, Archer Gong and Chen, Jiahua , year =. Gaussian. doi:10.1109/TIT.2023.3323346 , journal =
-
[51]
Sajedi, A. and Lawryshyn, Y. and Plataniotis, K. , year =. A. doi:10.1109/ICASSP49357.2023.10096094 , journal =
-
[52]
Characterisation of molecular motions in cryo-. eLife , author =. 2018 , note =. doi:10.7554/eLife.36861 , abstract =
-
[53]
Graphical Models and Image Processing , author =
Three-. Graphical Models and Image Processing , author =. 1997 , pages =. doi:10.1006/gmip.1997.0420 , abstract =
-
[54]
Journal of Molecular Biology , author =
Integrating. Journal of Molecular Biology , author =. 2023 , keywords =. doi:10.1016/j.jmb.2023.168014 , abstract =
-
[55]
Acta Crystallographica Section D: Structural Biology , author =
Protein identification from electron cryomicroscopy maps by automated model building and side-chain matching , volume =. Acta Crystallographica Section D: Structural Biology , author =. 2021 , note =. doi:10.1107/S2059798321001765 , abstract =
-
[56]
Nature Methods , author =. 2022 , note =. doi:10.1038/s41592-021-01389-9 , abstract =
-
[57]
Automated model building and protein identification in cryo-. Nature , author =. 2024 , note =. doi:10.1038/s41586-024-07215-4 , abstract =
-
[58]
Iterative model building, structure refinement and density modification with the. Acta Crystallographica. Section D, Biological Crystallography , author =. 2008 , pmid =. doi:10.1107/S090744490705024X , abstract =
-
[59]
Visualizing the Loss Landscape of Neural Nets
Li, Hao and Xu, Zheng and Taylor, Gavin and Studer, Christoph and Goldstein, Tom , year =. Visualizing the. doi:10.48550/ARXIV.1712.09913 , abstract =
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv.1712.09913
-
[60]
Structures of α-synuclein filaments from human brains with. Nature , author =. 2022 , pmid =. doi:10.1038/s41586-022-05319-3 , abstract =
-
[61]
Novel. Micromachines , author =. 2023 , pages =. doi:10.3390/mi14091674 , abstract =
-
[62]
Nature Methods , author =. 2024 , note =. doi:10.1038/s41592-024-02377-5 , abstract =
-
[63]
Journal of Structural Biology , author =
Fast multiscale reconstruction for. Journal of Structural Biology , author =. 2018 , keywords =. doi:10.1016/j.jsb.2018.09.008 , abstract =
-
[64]
Journal of Structural Biology , author =
Image formation modeling in cryo-electron microscopy , volume =. Journal of Structural Biology , author =. 2013 , keywords =. doi:10.1016/j.jsb.2013.05.008 , abstract =
-
[65]
doi:10.1101/2023.10.31.564872 , abstract =
Li, Yilai and Zhou, Yi and Yuan, Jing and Ye, Fei and Gu, Quanquan , month = nov, year =. doi:10.1101/2023.10.31.564872 , abstract =
-
[66]
Improving resolution and resolvability of single-particle. Nature Methods , author =. 2024 , note =. doi:10.1038/s41592-023-02082-9 , abstract =
-
[67]
Molecular biology of the cell
-
[68]
Nashed, Youssef S. G. and Poitevin, Frederic and Gupta, Harshit and Woollard, Geoffrey and Kagan, Michael and Yoon, Chun Hong and Ratner, Daniel , month = oct, year =. 2021. doi:10.1109/ICCVW54120.2021.00452 , urldate =
-
[69]
Levy, Axel and Poitevin, Frédéric and Martel, Julien and Nashed, Youssef and Peck, Ariana and Miolane, Nina and Ratner, Daniel and Dunne, Mike and Wetzstein, Gordon , month = oct, year =. Computer. doi:10.1007/978-3-031-19803-8_32 , abstract =
-
[70]
IEEE Transactions on Neural Networks and Learning Systems , author =
Machine. IEEE Transactions on Neural Networks and Learning Systems , author =. 2022 , note =. doi:10.1109/TNNLS.2021.3131325 , abstract =
-
[71]
Matter , author =. 2021 , keywords =. doi:10.1016/j.matt.2021.09.004 , abstract =
-
[72]
and Goncharov, A
Vainshtein, B. and Goncharov, A. , month = apr, year =. Determination of the spatial orientation of arbitrarily arranged identical particles of unknown structure from their projections , url =
-
[73]
Angular reconstitution:. Ultramicroscopy , author =. 1987 , pages =. doi:10.1016/0304-3991(87)90078-7 , abstract =
-
[74]
Analyzing
Alberts, Bruce and Johnson, Alexander and Lewis, Julian and Raff, Martin and Roberts, Keith and Walter, Peter , year =. Analyzing. Molecular
-
[75]
Journal of Structural Biology , author =
Deep generative modeling for volume reconstruction in cryo-electron microscopy , volume =. Journal of Structural Biology , author =. 2022 , pages =. doi:10.1016/j.jsb.2022.107920 , language =
-
[76]
IEEE Signal Processing Magazine , author =
Single-. IEEE Signal Processing Magazine , author =. 2020 , pages =. doi:10.1109/MSP.2019.2957822 , number =
-
[77]
Ghanem, Karam and Bzdok, Danilo , month = feb, year =. The. doi:10.48550/arXiv.2402.13369 , abstract =
-
[78]
, month = feb, year =
Song, Yang and Kingma, Diederik P. , month = feb, year =. How to
-
[79]
Chang, Ziyi and Koulieris, George Alex and Shum, Hubert P. H. , month = oct, year =. On the
-
[80]
https://arxiv.org/pdf/2306.04542 , url =
-
[81]
Score-Based Generative Modeling through Stochastic Differential Equations
Song, Yang and Sohl-Dickstein, Jascha and Kingma, Diederik P. and Kumar, Abhishek and Ermon, Stefano and Poole, Ben , year =. Score-. doi:10.48550/ARXIV.2011.13456 , abstract =
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv.2011.13456 2011
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.