EasyNano: rapid epitope-targeted nanobody CDR design via differentiable distogram optimization with ESMFold2
Pith reviewed 2026-06-27 05:24 UTC · model grok-4.3
The pith
EasyNano optimizes nanobody CDR sequences by gradient descent on ESMFold2 distance predictions to target user-specified epitopes.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
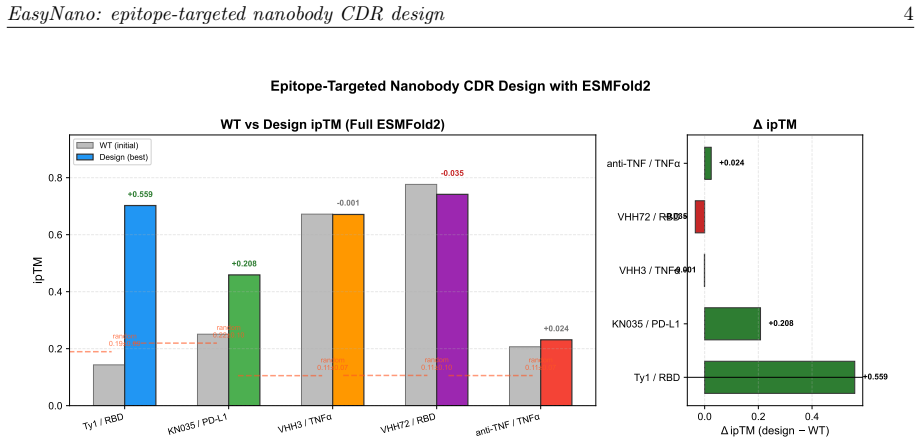
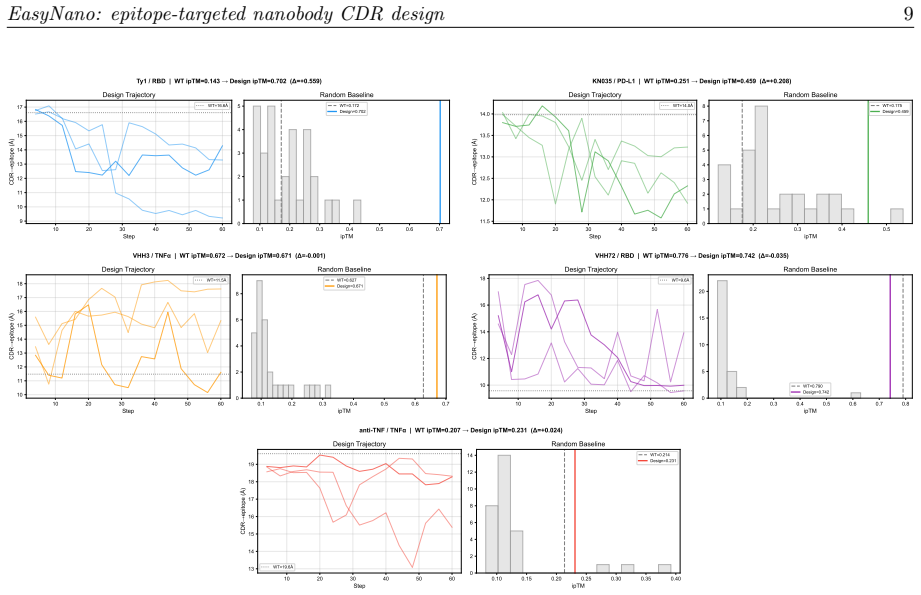
EasyNano optimizes CDR residue logits via gradient descent through the ESMFold2 pairwise distance distogram, using the lightweight ESMFold2-Fast model as a differentiable oracle guided by a composite loss including a dedicated epitope proximity term. A full ESMFold2 CA-coordinate structure prior prevents framework pose drift. Across six target-framework pairs the procedure improves ipTM by up to +0.559 while preserving ipTM on already-strong binders.
What carries the argument
Differentiable optimization of CDR logits through the ESMFold2 distogram, driven by a composite loss that includes an epitope proximity term.
If this is right
- Designed CDRs reach statistically higher ipTM than random sequences drawn from the same length distribution.
- Framework geometry remains inside the native pose basin after optimization, as confirmed by Kabsch alignment to crystal structures.
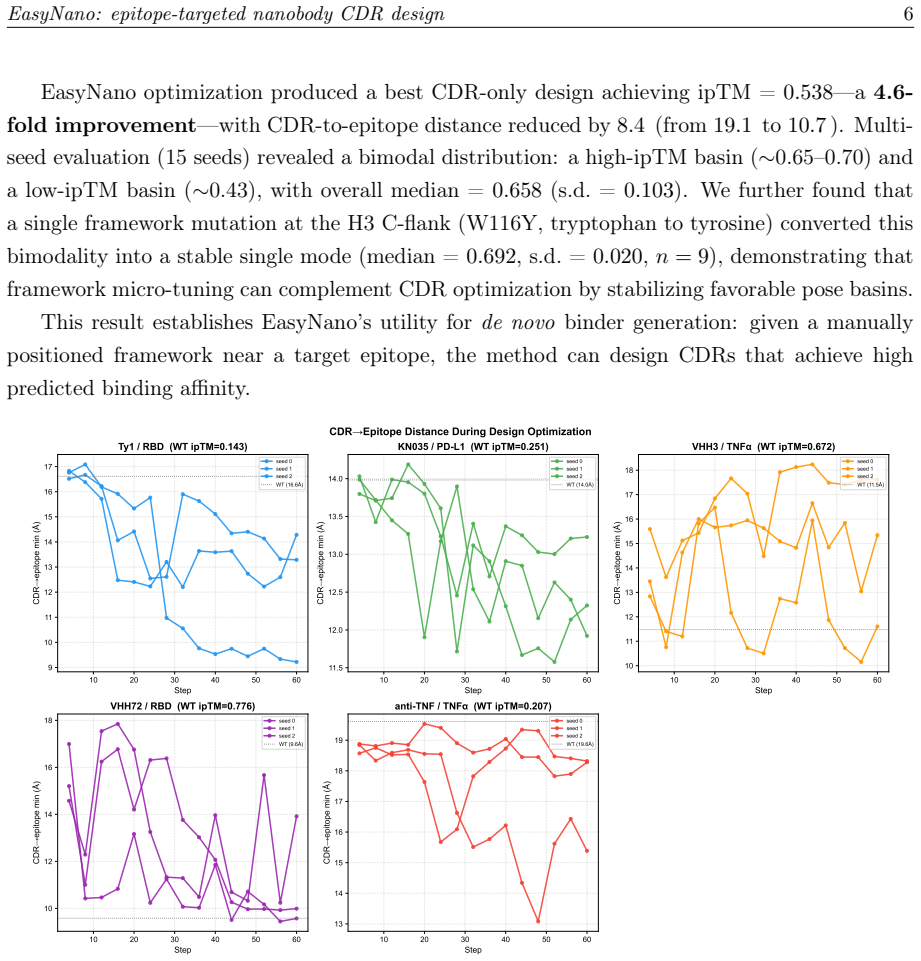
- Multiple random seeds produce distinct local minima, indicating that replicate runs increase the chance of finding good solutions.
- The same pipeline works for both recovering known binders and designing new ones against manually docked epitopes.
- The lightweight ESMFold2-Fast model can serve as a fast, differentiable surrogate while the larger model supplies the pose prior.
Where Pith is reading between the lines
- If the ipTM gains translate to experiment, the method could shorten the computational phase of nanobody campaigns from days to minutes and allow more epitope choices to be explored.
- The dependence on replicate runs suggests that future versions might benefit from explicit diversity penalties or ensemble losses to reduce the number of trials needed.
- Because the loss is built around a structure predictor rather than a sequence-only model, the same differentiable-oracle pattern could be tested on other loop-design problems such as antibody or peptide engineering.
- The emergence of wild-type logit bias as a tunable knob for mutability points to a practical control that may generalize to other gradient-based protein design tasks.
Load-bearing premise
ipTM scores computed by ESMFold2 on the optimized sequences will correspond to real binding and stability once the nanobodies are made and tested.
What would settle it
Laboratory measurement of binding affinity or stability for the designed nanobodies against their intended epitopes, compared with the ipTM values reported by the method.
Figures


read the original abstract
Computational design of nanobodies that bind user-specified protein epitopes could transform therapeutic development, but current methods either rely on stochastic sampling requiring days of GPU computation or inverse folding approaches unable to target epitopes directly. Here we present EasyNano, a practical pipeline for rapid, epitope-targeted nanobody complementarity-determining region (CDR) design that operates in approximately 10-20 minutes on a high-end personal workstation. EasyNano optimizes CDR residue logits via gradient descent through the ESMFold2 pairwise distance distogram, using the lightweight ESMFold2-Fast model (721M) as a differentiable oracle guided by a composite loss including a dedicated epitope proximity term. A full ESMFold2 (1.3B) CA-coordinate structure prior prevents framework pose drift. The wild-type logit initialization bias emerges as a critical practical parameter controlling CDR mutability. Across six target-framework pairs spanning self-recovery and de novo design scenarios, EasyNano improves ipTM by up to +0.559 -- from 0.143 to 0.702 (Ty1/RBD) -- and achieves a 4.6-fold improvement (ipTM 0.117 to 0.538) on a manually docked AQP4-targeting framework, while preserving ipTM on already-strong binders. Random CDR baselines (n=30 per target) confirm statistical significance (5.7 sigma above random mean for Ty1). Multi-seed analysis reveals diverse local minima, underscoring the importance of replicate runs. Kabsch cross-validation against crystal structures confirms that designed CDRs preserve the framework pose basin. EasyNano demonstrates that ESMFold2-based differentiable optimization provides a fast, practical, and epitope-specific approach to nanobody CDR design.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces EasyNano, a pipeline for rapid epitope-targeted nanobody CDR design. It optimizes CDR residue logits via gradient descent through the ESMFold2 pairwise distance distogram using the lightweight ESMFold2-Fast model as a differentiable oracle, guided by a composite loss with a dedicated epitope proximity term and a full ESMFold2 CA-coordinate prior to prevent framework drift. Across six target-framework pairs, it reports ipTM gains of up to +0.559 (e.g., Ty1/RBD from 0.143 to 0.702), a 4.6-fold improvement on a de-novo AQP4 case, statistical significance over n=30 random baselines, multi-seed diversity, and Kabsch-validated pose preservation, positioning the method as practical (10-20 min on a workstation).
Significance. If the reported ipTM gains were shown to correspond to actual binding and stability, EasyNano would offer a fast, epitope-specific computational design tool that addresses limitations of stochastic sampling or inverse-folding methods. The differentiable-oracle approach, explicit epitope term, and multi-seed analysis are constructive elements. However, the complete dependence on in silico metrics from the optimization model itself, without orthogonal predictors or experimental data, substantially limits the current significance for therapeutic nanobody development.
major comments (3)
- [Abstract] Abstract: the central claim that EasyNano achieves epitope-targeted CDR design is supported solely by ipTM scores produced by the same ESMFold2 family used as the optimization oracle; no independent structure predictor, orthogonal metric, or wet-lab binding/stability data is provided to show that the +0.559 ipTM gains (or the 4.6-fold AQP4 improvement) reflect functional sequences rather than optimization artifacts.
- [Abstract] Abstract: the statistical-significance statement (5.7 sigma above random mean for Ty1) rests on n=30 random CDR baselines, yet the manuscript supplies no description of how those baselines were sampled or whether they were subjected to the same epitope-proximity and pose-preservation constraints, undermining the cross-target claim of reliable improvement.
- [Abstract] Abstract: for the de-novo AQP4 case the reported ipTM rise from 0.117 to 0.538 is presented as evidence of successful epitope targeting, but the only supporting evidence is the self-reported ipTM; no cross-check against an independent folding model or assessment of whether the optimized CDRs actually contact the intended epitope residues is given.
minor comments (2)
- [Abstract] The wild-type logit initialization bias is identified as a critical practical parameter, yet its precise functional form and sensitivity analysis are not detailed enough for independent reproduction.
- Notation: 'ESMFold2' and 'ESMFold2-Fast (721M)' should be explicitly mapped to the publicly released model versions and parameter counts to avoid ambiguity.
Simulated Author's Rebuttal
We thank the referee for the detailed and constructive review. We respond point-by-point to the major comments, acknowledging the in silico nature of the validation while clarifying methodological details and offering targeted revisions where feasible.
read point-by-point responses
-
Referee: [Abstract] Abstract: the central claim that EasyNano achieves epitope-targeted CDR design is supported solely by ipTM scores produced by the same ESMFold2 family used as the optimization oracle; no independent structure predictor, orthogonal metric, or wet-lab binding/stability data is provided to show that the +0.559 ipTM gains (or the 4.6-fold AQP4 improvement) reflect functional sequences rather than optimization artifacts.
Authors: We agree that all quantitative claims rely on ipTM and related metrics from the ESMFold2 family, with optimization performed via the Fast variant and evaluation using the full model. This constitutes a self-consistent in silico demonstration rather than orthogonal or experimental validation. The abstract frames EasyNano as a computational pipeline, and relative gains versus random and wild-type baselines are intended to show the optimization procedure's effectiveness within that framework. We do not claim functional binding. We will add a clarifying sentence in the abstract and discussion noting the in silico scope and absence of experimental data. revision: partial
-
Referee: [Abstract] Abstract: the statistical-significance statement (5.7 sigma above random mean for Ty1) rests on n=30 random CDR baselines, yet the manuscript supplies no description of how those baselines were sampled or whether they were subjected to the same epitope-proximity and pose-preservation constraints, undermining the cross-target claim of reliable improvement.
Authors: The current text states that random CDR baselines were generated but does not detail the sampling distribution or whether the full composite loss (including epitope and pose terms) was applied during their evaluation. We will revise the methods section to specify the exact sampling procedure (e.g., uniform amino-acid sampling within CDR lengths) and confirm that baselines receive the same ipTM evaluation protocol, thereby allowing direct comparison under equivalent constraints. revision: yes
-
Referee: [Abstract] Abstract: for the de-novo AQP4 case the reported ipTM rise from 0.117 to 0.538 is presented as evidence of successful epitope targeting, but the only supporting evidence is the self-reported ipTM; no cross-check against an independent folding model or assessment of whether the optimized CDRs actually contact the intended epitope residues is given.
Authors: The AQP4 result is driven by the explicit epitope-proximity term in the loss, which directly penalizes distance to the target epitope residues. We will add the final epitope-proximity loss values and a short contact-map analysis for this case in the results section. An independent folding model evaluation is not currently performed; we can note this limitation and, if space permits, report a limited cross-check with an alternative predictor if computationally feasible in revision. revision: partial
- Provision of wet-lab binding or stability data, which is absent from the current computational study and cannot be supplied without new experiments.
Circularity Check
ipTM gains measured on same ESMFold2 oracle used for optimization, no orthogonal validation
full rationale
The paper presents a method that performs gradient descent on CDR logits using ESMFold2-Fast as a differentiable oracle with a composite loss containing an epitope proximity term, then reports ipTM improvements (up to +0.559) evaluated on the ESMFold2 family. No load-bearing self-citation, self-definitional loop, or fitted-input-called-prediction is exhibited: the pretrained model is external, the optimization objective is not shown to be identical to the reported ipTM metric by the paper's own equations, and controls (random baselines, multi-seed, Kabsch) address sampling but do not create a circular reduction. This is a standard use of an external ML oracle; the result is not forced by construction to the paper's inputs. Score kept low per rules for self-contained external-model usage without explicit reduction.
Axiom & Free-Parameter Ledger
free parameters (1)
- wild-type logit initialization bias
axioms (1)
- domain assumption ESMFold2-Fast and full ESMFold2 produce sufficiently accurate pairwise distances and CA coordinates to serve as optimization oracles for designed sequences.
Reference graph
Works this paper leans on
-
[1]
Nanobodies: natural single-domain antibodies.Annu
Muyldermans, S. Nanobodies: natural single-domain antibodies.Annu. Rev. Biochem.82, 775–797 (2013)
2013
-
[2]
Hamers-Casterman, C.et al.Naturally occurring antibodies devoid of light chains.Nature 363, 446–448 (1993)
1993
-
[3]
& Muyldermans, S
Jovčevska, I. & Muyldermans, S. The therapeutic potential of nanobodies.BioDrugs34, 11–26 (2020)
2020
-
[4]
Scully, M.et al.Caplacizumab treatment for acquired thrombotic thrombocytopenic pur- pura.N. Engl. J. Med.380, 335–346 (2019)
2019
-
[5]
Bioin- form.21, 1549–1567 (2020)
Norman, R.A.et al.Computationalapproachestotherapeuticantibodydesign.Brief. Bioin- form.21, 1549–1567 (2020)
2020
-
[6]
Jumper, J.et al.Highly accurate protein structure prediction with AlphaFold.Nature596, 583–589 (2021)
2021
-
[7]
Baek, M.et al.Accurate prediction of protein structures and interactions using a three-track neural network.Science373, 871–876 (2021)
2021
-
[8]
Lin, Z.et al.Evolutionary-scale prediction of atomic-level protein structure with a language model.Science379, 1123–1130 (2023)
2023
-
[9]
Abramson, J.et al.Accurate structure prediction of biomolecular interactions with Al- phaFold 3.Nature630, 493–500 (2024)
2024
-
[10]
Science378, 49–56 (2022)
Dauparas, J.et al.Robustdeeplearning–basedproteinsequencedesignusingProteinMPNN. Science378, 49–56 (2022)
2022
-
[11]
ICML (2022)
Hsu, C.et al.Learning inverse folding from millions of predicted structures.Proc. ICML (2022)
2022
-
[12]
Anishchenko, I.et al.De novo protein design by deep network hallucination.Nature600, 547–552 (2021)
2021
-
[13]
Watson, J.L.et al.De novo design of protein structure and function with RFdiffusion.Nature 620, 1089–1100 (2023)
2023
-
[14]
Wang, J.et al.Scaffolding protein functional sites using deep learning.Science377, 387–394 (2022)
2022
-
[15]
Commun.14, 2389 (2023)
Ruffolo, J.A.et al.Fast, accurate antibody structure prediction from deep learning on massive set of natural antibodies.Nat. Commun.14, 2389 (2023)
2023
-
[16]
EasyNano: epitope-targeted nanobody CDR design14
Ruffolo, J.A., Gray, J.J.&Sulam, J.Decipheringantibodyaffinitymaturationwithlanguage models and weakly supervised learning.arXiv(2021). EasyNano: epitope-targeted nanobody CDR design14
2021
-
[17]
& Deane, C.M
Olsen, T.H., Moal, I.H. & Deane, C.M. AbLang: an antibody language model for completing antibody sequences.Bioinform. Adv.2, vbac046 (2022)
2022
-
[18]
Neural Inf
Luo, S.et al.Antigen-specific antibody design and optimization with diffusion-based gener- ative models for protein structures.Adv. Neural Inf. Process. Syst.35, 9754–9767 (2022)
2022
-
[19]
& Liu, Y
Kong, X., Huang, W. & Liu, Y. End-to-end full-atom antibody design.Proc. ICML(2023)
2023
-
[20]
ICLR(2022)
Jin, W.et al.Iterative refinement graph neural network for antibody sequence-structure co-design.Proc. ICLR(2022)
2022
-
[21]
Commun.11, 4420 (2020)
Hanke, L.et al.An alpaca nanobody neutralizes SARS-CoV-2 by blocking receptor inter- action.Nat. Commun.11, 4420 (2020)
2020
-
[22]
Commun.12, 4003 (2021)
Wang, Y.et al.A potent neutralizing nanobody against SARS-CoV-2.Nat. Commun.12, 4003 (2021)
2021
-
[23]
Cell Discov.5, 43 (2019)
Zhang, F.et al.StructuralbasisofanovelPD-L1nanobodyforimmunecheckpointblockade. Cell Discov.5, 43 (2019)
2019
-
[24]
He, B.L.et al.A protein language model for all domains of life.bioRxiv(2022)
2022
-
[25]
Rives, A.et al.Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences.Proc. Natl. Acad. Sci.118, e2016239118 (2021)
2021
-
[26]
Liu, Y.et al.De novo design of programmable protein interactions.Nat. Rev. Bioeng.(2024)
2024
-
[27]
Plückthun, A.Designedankyrinrepeatproteins(DARPins).Annu. Rev. Pharmacol. Toxicol. 55, 489–511 (2015)
2015
-
[28]
Koide, A.et al.Teaching an old scaffold new tricks: monobodies.J. Mol. Biol.415, 393–405 (2012)
2012
-
[29]
Kunzmann, P.et al.Biotite: a unifying open source computational biology framework in Python.BMC Bioinform.24, 346 (2023)
2023
-
[30]
& Chothia, C
Al-Lazikani, B., Lesk, A.M. & Chothia, C. Standard conformations for the canonical struc- tures of immunoglobulins.J. Mol. Biol.273, 927–948 (1997)
1997
-
[31]
(this work, internal analysis)
Hu, Y.et al.ESMFold2 predicted complex pose is determined by framework sequence, not diffusion initialization. (this work, internal analysis)
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.