MGOS: A Library for Molecular Geometry and its Operating System
Pith reviewed 2026-05-25 13:29 UTC · model grok-4.3
The pith
Molecular Geometry framework lets researchers model atomic arrangements using volume, area, and other standard measures, implemented through the MGOS library of callable functions.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Molecular Geometry (MG) is a theoretical framework for the geometry of atomic arrangement that expresses complicated molecular structure problems in terms of standard notions such as volume and area. MGOS consists of callable functions that realize the MG theory, allowing these functions to be embedded in application programs. This combination supplies accurate solutions to geometric queries involving atomic arrangements and frees users from the task of developing geometric algorithms.
What carries the argument
The MG framework, which recasts molecular structure problems as calculations over volume, area, and related geometric primitives, realized through the MGOS set of callable functions.
If this is right
- MG simplifies the modeling of molecular structure problems to elementary geometric notions.
- MGOS functions embed directly into application programs for accurate geometric query solutions.
- Researchers avoid the development and implementation of geometric algorithms.
- Use of MGOS for spherical entities parallels the role of math libraries in general-purpose scientific programming.
Where Pith is reading between the lines
- Standardization around MG primitives could reduce duplication of geometric code across separate molecular modeling projects.
- The spherical-entity focus leaves open whether the same reduction to volume and area extends cleanly to non-spherical molecular components.
- Embedding MGOS in existing simulation packages might allow direct comparison of geometric accuracy against hand-coded alternatives.
Load-bearing premise
No general framework of mathematical or computational theory for the geometry of atomic arrangement already exists.
What would settle it
A documented prior framework that already permits modeling of atomic arrangements through volume, area, and similar standard measures without requiring users to develop custom geometric algorithms.
Figures




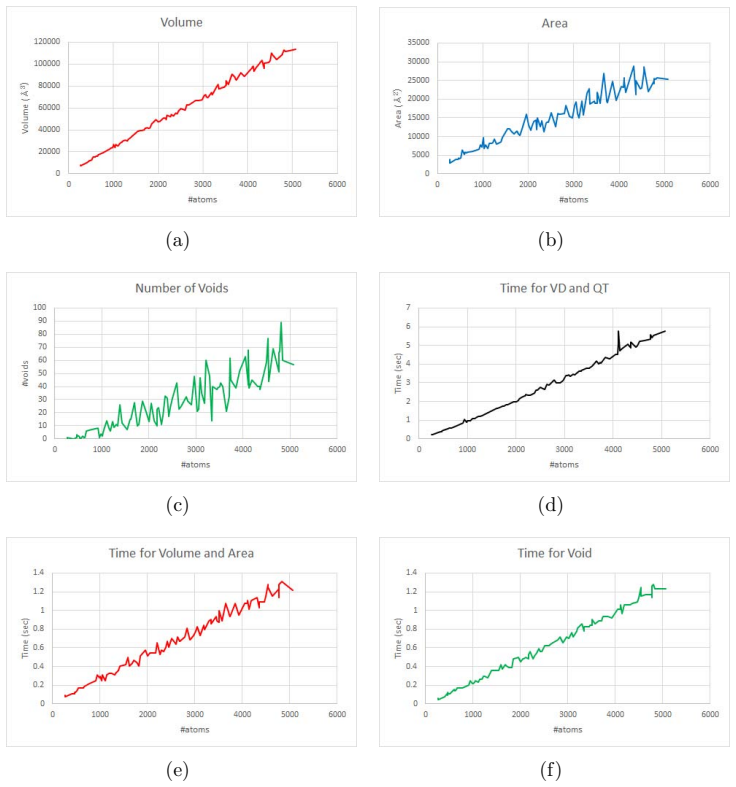
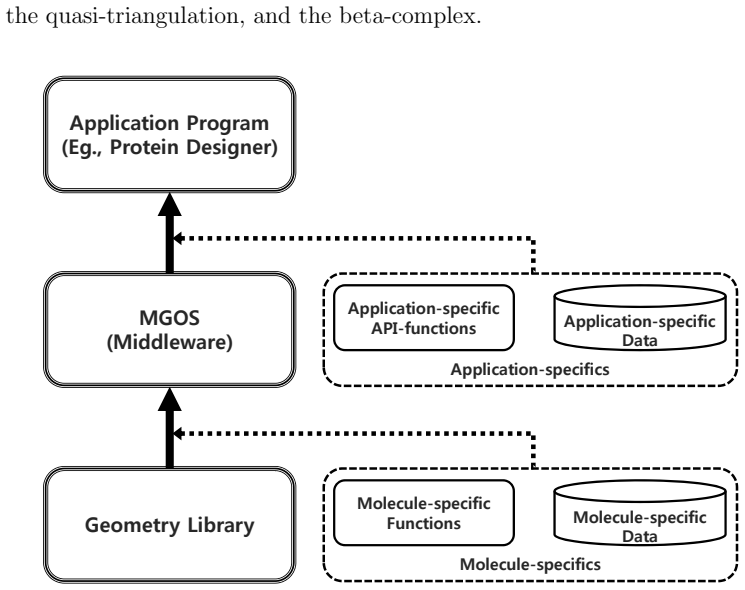
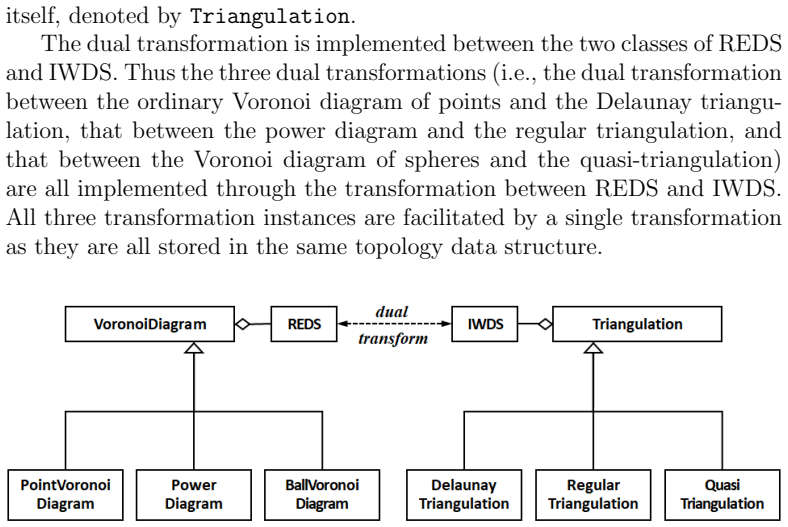
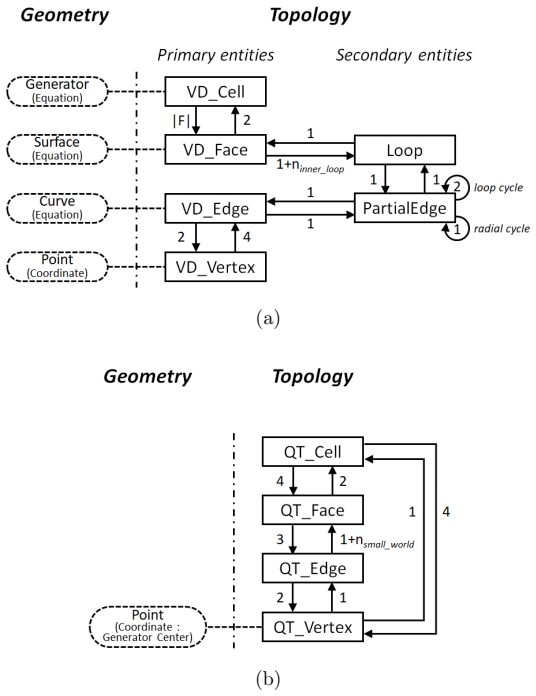
read the original abstract
The geometry of atomic arrangement underpins the structural understanding of molecules in many fields. However, no general framework of mathematical/computational theory for the geometry of atomic arrangement exists. Here we present "Molecular Geometry (MG)" as a theoretical framework accompanied by "MG Operating System (MGOS)" which consists of callable functions implementing the MG theory. MG allows researchers to model complicated molecular structure problems in terms of elementary yet standard notions of volume, area, etc. and MGOS frees them from the hard and tedious task of developing/implementing geometric algorithms so that they can focus more on their primary research issues. MG facilitates simpler modeling of molecular structure problems; MGOS functions can be conveniently embedded in application programs for the efficient and accurate solution of geometric queries involving atomic arrangements. The use of MGOS in problems involving spherical entities is akin to the use of math libraries in general purpose programming languages in science and engineering.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper presents 'Molecular Geometry (MG)' as a new theoretical framework for modeling the geometry of atomic arrangements in molecules using elementary notions such as volume and area, accompanied by the 'MG Operating System (MGOS)' library consisting of callable functions that implement MG theory. It claims this approach simplifies molecular structure problems and eliminates the need for researchers to develop geometric algorithms, positioning MGOS as a foundational library akin to math libraries in general-purpose programming.
Significance. If the MG framework proves general and the MGOS implementations are efficient and accurate for queries involving atomic (spherical) arrangements, the work could provide a useful standardized interface for geometric computations in computational chemistry and structural biology, reducing redundant implementation effort across applications.
major comments (1)
- [Abstract] Abstract: The central premise that 'no general framework of mathematical/computational theory for the geometry of atomic arrangement exists' is stated without citations, literature review, or explicit comparison to prior computational geometry tools for molecules (such as those based on Voronoi diagrams, alpha shapes, or existing molecular surface libraries). This assertion is load-bearing for the claimed novelty and necessity of both MG and MGOS.
minor comments (1)
- [Abstract] The abstract refers to 'spherical entities' and 'atomic arrangement' but provides no explicit scope, assumptions, or limitations of the MG framework.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback. We address the single major comment below.
read point-by-point responses
-
Referee: [Abstract] Abstract: The central premise that 'no general framework of mathematical/computational theory for the geometry of atomic arrangement exists' is stated without citations, literature review, or explicit comparison to prior computational geometry tools for molecules (such as those based on Voronoi diagrams, alpha shapes, or existing molecular surface libraries). This assertion is load-bearing for the claimed novelty and necessity of both MG and MGOS.
Authors: We agree that the abstract states the absence of a general framework without citations or comparisons, and that this claim supports the novelty argument. The full manuscript develops MG as a framework centered on elementary geometric measures (volume, area) together with an OS-style library interface; however, the abstract itself does not reference existing molecular computational-geometry literature. In the revised manuscript we will shorten and qualify the claim in the abstract, add a brief clause acknowledging Voronoi-diagram and alpha-shape approaches, and indicate how MGOS differs by supplying a standardized callable interface rather than requiring per-application algorithm development. revision: yes
Circularity Check
No circularity: library description with no derivations or self-referential reductions
full rationale
The paper presents a software library (MGOS) implementing a conceptual framework (MG) for molecular geometry but contains no equations, derivations, fitted parameters, or mathematical claims that could reduce to inputs by construction. The central premise that no prior general framework exists is an unsubstantiated novelty assertion rather than a load-bearing derivation step; per the analysis rules this falls under correctness/novelty risk, not circularity. No self-citations, ansatzes, or uniqueness theorems are invoked in a way that creates the enumerated circular patterns. The work is self-contained as a descriptive library contribution.
Axiom & Free-Parameter Ledger
invented entities (1)
-
Molecular Geometry (MG) framework
no independent evidence
Reference graph
Works this paper leans on
- [1]
-
[2]
H. Li, M. Eddaoudi, M. O’Keeffe, O. M. Yaghi, Design and synthesis of an exceptionally stable and highly porous metal-organic framework, Nature 402 (1999) 276–279
work page 1999
-
[3]
H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. Yazaydin, R. Q.Snurr, M. OKeeffe, Ultrahigh porosity in metal- organic frameworks, Science 329 (5990) (2010) 424–428
work page 2010
-
[4]
Chothia, Hydrophobic bonding and accessible surface area in pro- teins, Nature 248 (1974) 338–339
C. Chothia, Hydrophobic bonding and accessible surface area in pro- teins, Nature 248 (1974) 338–339
work page 1974
-
[5]
M. L. Connolly, Solvent-accessible surfaces of proteins and nucleic acids, Science 221 (1983) 709–713
work page 1983
-
[6]
S. J. Hubbard, K.-H. Gross, P. Argos, Intramolecular cavities in globular proteins, Protein Engineering 7 (5) (1994) 613–626
work page 1994
-
[7]
D. A. Doyle, joao Morais Cabral, R. A. Pfuetzer, A. Kuo, J. M. Gulbis, S. L. Cohan, B. T. Chait, R. Mackinnon, The structure of the potassium channel: Molecular basis of k conduction and selectivity, Science 280 (1998) 69–77
work page 1998
- [8]
-
[9]
X. Liu, E. C.Theil, Ferritins: Dynamic management of biological iron oxygen chemistry, Accounts of Chemical Research 38 (3) (2005) 167–175
work page 2005
-
[10]
C. H. Park, S. Y. Lee, D. S. Hwang, D. W. Shin, D. H. Cho, K. H. Lee, T.-W. Kim, T.-W. Kim, M. Lee, D.-S. Kim, C. M. Doherty, A. W. Thornton, A. J. Hill, M. D. Guiver, Y. M. Lee, Nano-crack regulated self-humidifying membranes, Nature 532 (7600) (2016) 480–483
work page 2016
-
[11]
T. Gerling, K. F. Wagenbauer, A. M. Neuner, H. Dietz, Dynamic dna devices and assemblies formed by shape-complementary, nonbase pairing 3d components, Science 347 (6229) (2015) 1446–1452
work page 2015
-
[12]
M. N. O’Brien, M. R. Jones, B. Lee, C. A. Mirkin, Anisotropic nanopar- ticle complementarity in DNA-mediated co-crystallization, Nature Ma- terials 14 (2015) 833–839
work page 2015
-
[13]
E. Fischer, Synthesen in der zuckergruppe, Berichte der deutschen chemischen Gesellschaft 23 (2) (1890) 2114–2141
- [14]
- [15]
-
[16]
G. J. Kleywegt, T. A. Jones, Detection, delineation, measurement and display of cavities in macromolecular structures, Acta Crystallographica Section D 50 (1994) 178–185
work page 1994
-
[17]
F. M. Richards, Areas, volumes, packing, and protein structure, Annual Review of Biophysics and Bioengineering 6 (1977) 151–176
work page 1977
-
[18]
J.-K. Kim, Y. Cho, R. A. Laskowski, S. E. Ryu, K. Sugihara, D.-S. Kim, BetaVoid: molecular voids via beta-complexes and Voronoi diagrams, Proteins: Structure, Functions, and Bioinformatics 82 (9) (2014) 1829– 1849
work page 2014
-
[19]
J.-K. Kim, Y. Cho, M. Lee, R. A. Laskowski, S. E. Ryu, K. Sugihara, D.- S. Kim, BetaCavityWeb: a webserver for molecular voids and channels, Nucleic Acids Research 43 (W1) (2015) W413–W418. 28
work page 2015
-
[20]
RCSB Protein Data Bank, http://www.rcsb.org/pdb/
- [21]
-
[22]
S. Horiuchi, H. Tanaka, E. Sakuda, Y. Arikawa, K. Umakoshi, Encapsu- lation condition dependent photophysical properties of polypyridyl ru(ii) complexes within a hydrogen-bonded capsule, Dalton Transactions 48 (2019) 5156–5160
work page 2019
-
[23]
A. Shimada, M. Kubo, S. Baba, K. Yamashita, K. Hirata, G. Ueno, T. Nomura, T. Kimura, K. Shinzawa-Itoh, J. Baba, K. Hatano, Y. Eto, A. Miyamoto, H. Murakami, T. Kumasaka, S. Owada, K. Tono, M. Yabashi, Y. Yamaguchi, S. Yanagisawa, M. Sakaguchi, T. Ogura, R. Komiya, J. Yan, E. Yamashita, M. Yamamoto, H. Ago, S. Yoshikawa, T. Tsukihara, A nanosecond time-re...
work page 2017
-
[24]
L. Ducassoua, L. Dhers, G. Jonasson, N. Pietrancosta, J.-L. Boucher, D. Mansuy, F. Andr´ e, Membrane-bound human orphan cytochrome P450 2U1: Sequence singularities, construction of a full 3d model, and substrate docking, Biochimie 140 (2017) 166–175
work page 2017
- [25]
-
[26]
F. J. Rizzuto, J. R. Nitschke, Stereochemical plasticity modulates coop- erative binding in a Co II 12L6 cuboctahedron, Nature Chemistry 9 (2017) 903–908
work page 2017
-
[27]
G. Markiewicz, A. Jenczak, M. Ko lodziejski, J. J. Holstein, J. K. M. Sanders, A. R. Stefankiewicz, SelectiveC70 encapsulation by a robust oc- tameric nanospheroid held together by 48 cooperative hydrogen bonds, Nature Communications 8 (15109) (2017) 1–8
work page 2017
-
[28]
V. Chaptal, F. Delolme, A. Kilburg, S. Magnard, C. Montigny, M. Pi- card, C. Prier, L. Monticelli, O. Bornert, M. Agez, S. Ravaud, C. Orelle, 29 R. Wagner, A. Jawhari, I. Broutin, E. Pebay-Peyroula, J.-M. Jault, H. R. Kaback, M. le Maire, P. Falson, Quantification of detergents com- plexed with membrane proteins, Scientific Reports 7 (41751) (2017) 1– 12
work page 2017
- [29]
-
[30]
S. Kitanovic, C. A. Marks-Fife, Q. A. Parkes, P. R. Wilderman, J. R. Halpert, M. D. Dearing, Cytochrome p450 2b diversity in a dietary spe- cialistthe red tree vole (arborimus longicaudus), Journal of Mammalogy 99 (3) (2018) 578–585
work page 2018
- [31]
-
[32]
B. Htan, D. Luo, C. Ma, J. Zhang, Q. Gan, Water-trapping of metal- organic cages with endohedral variation, Crystal Growth & Design 19 (5) (2019) 2862–2868
work page 2019
-
[33]
A. Chakravorty, E. Gallicchio, A gridbased algorithm in conjunction with a gaussianbased model of atoms for describing molecular geometry, Computational Chemistry 40 (12) (2019) 1290–1304
work page 2019
-
[34]
D.-S. Kim, Y. Cho, D. Kim, Euclidean Voronoi diagram of 3D balls and its computation via tracing edges, Computer-Aided Design 37 (13) (2005) 1412–1424
work page 2005
-
[35]
D.-S. Kim, D. Kim, Y. Cho, K. Sugihara, Quasi-triangulation and in- terworld data structure in three dimensions, Computer-Aided Design 38 (7) (2006) 808–819
work page 2006
-
[36]
D.-S. Kim, Y. Cho, K. Sugihara, Quasi-worlds and quasi-operators on quasi-triangulations, Computer-Aided Design 42 (10) (2010) 874–888
work page 2010
-
[37]
D.-S. Kim, Y. Cho, K. Sugihara, J. Ryu, D. Kim, Three-dimensional beta-shapes and beta-complexes via quasi-triangulation, Computer- Aided Design 42 (10) (2010) 911–929. 30
work page 2010
-
[38]
J. A. Le Bel, On the relations which exist between the atomic formulas of organic compounds and the rotatory power of their solutions (orig. publ. 1874), in: G. M. Richardson (Ed.), The Foundations of Stereochemistry, 1901, pp. 47–59, the Richardson translations have been reprinted in Benfey, O.T., Ed. Classics in the Theory of Chemical Combination; Dover: New
work page 1901
-
[39]
J. H. van’t Hoff, A suggestion looking to the extension into space of the structural formulas at present used in chemistry, and a note upon the relations between the optical activity and the chemical constitution of organic compounds (orig. publ. 1874), in: G. M. Richardson (Ed.), The Foundations of Stereochemistry, 1901, pp. 66–73
work page 1901
-
[40]
R. B. Grossman, Van’t hoff, le bel, and the development of stereochem- istry: A reassessment, Journal of chemical education 66 (1) (1989) 30– 33
work page 1989
-
[41]
N. Sidgwick, H. Powell, Bakerian lecture. stereochemical types and va- lency groups, in: Proceedings of the Royal Society A: Mathematical, Physical & Engineering Sciences, Vol. 176, 1940
work page 1940
-
[42]
R. J. Gillespie, Molecular Geometry, Van Nostrand Reinhold, 1972
work page 1972
-
[43]
R. J. Gillespie, R. S. Nyholm, Inorganic stereochemistry, Quarterly Re- views, Chemical Society 11 (4) (1957) 339–380
work page 1957
-
[44]
R. J. Gillespie, The valence-shell electron-pair repulsion (VSEPR) the- ory of directed valency, Journal of chemical education 40 (6) (1963) 295–301
work page 1963
-
[45]
R. Gillespie, Electron correlation and molecular shape, Canadian Jour- nal of Chemistry 38 (1960) 818–826
work page 1960
-
[46]
E. Fischer, Syntheses in the purine and sugar group, Nobel Lectures in Chemistry 1901-1921, Elsevier, Amsterdam, 1966
work page 1901
-
[47]
Crick, The packing of a-Helices: Simple coiled-coils, Acta Crystallo- graphica 6 (1953) 689–697
F. Crick, The packing of a-Helices: Simple coiled-coils, Acta Crystallo- graphica 6 (1953) 689–697
work page 1953
- [48]
- [49]
-
[50]
J. C. Kendrew, R. E. Dickerson, B. E. Strandberg, R. G. Hart, D. R. Davies, Structure of myoglobin: A three-dimensional fourier synthesis at 2 ˚ a. resolution, Nature 185 (4711) (1960) 422–427
work page 1960
-
[51]
B. Lee, F. M. Richards, The interpretation of protein structures: Es- timation of static accessibility, Journal of Molecular Biology 55 (1971) 379–400
work page 1971
-
[52]
Chothia, Structural invariants in protein folding, Nature 254 (27) (1975) 304–308
C. Chothia, Structural invariants in protein folding, Nature 254 (27) (1975) 304–308
work page 1975
-
[53]
F. M. Richards, The interpretation of protein structures: Total volume, group volume distributions and packing density, Journal of Molecular Biology 82 (1974) 1–14
work page 1974
-
[54]
R. A. Laskowski, M. W. MacArthur, D. S. Moss, J. M. Thornton, PROCHECK: a program to check the stereochemical quality of protein structures, Journal of Applied Crystallography 26 (1993) 283–291
work page 1993
-
[55]
V. B. Chen, W. B. A. III, J. J. Headd, D. A. Keedy, R. M. Immormino, G. J. Kapral, L. W. Murray, J. S. Richardson, D. C. Richardson, Mol- Probity: All-atom structure validation for macromolecular crystallogra- phy, Acta Crystallographica Section D: Biological Crystallography 66 (1) (2010) 12–21
work page 2010
-
[56]
I. W. Davis, A. Leaver-Fay, V. B. Chen, J. N. Block, G. J. Kapral, X. Wang, L. W. Murray, W. B. A. III, J. Snoeyink, J. S. Richardson, D. C. Richardson, MolProbity: all-atom contacts and structure valida- tion for proteins and nucleic acids, Nucleic Acids Research 35 (2007) W375–383
work page 2007
-
[57]
W.-H. Shin, J.-K. Kim, D.-S. Kim, C. Seok, GalaxyDock2: protein- ligand docking using beta-complex and global optimization, Journal of Computational Chemistry 34 (30) (2013) 2647–2656
work page 2013
-
[58]
J. Ryu, M. Lee, J. cha, R. A. Laskowski, S. E. Ryu, D.-S. Kim, Be- taSCPWeb: side-chain prediction for protein structures using voronoi 32 diagrams and geometry prioritization, Nucleic Acids Research 44 (W1) (2016) W416–W423
work page 2016
-
[59]
Bondi, van der Waals volumes and radii, Journal of Physical Chem- istry 68 (1964) 441–451
A. Bondi, van der Waals volumes and radii, Journal of Physical Chem- istry 68 (1964) 441–451
work page 1964
-
[60]
J. C. Slater, Atomic radii in crystals, Journal of Chemical Physics 41 (10) (1964) 3199–3204
work page 1964
-
[61]
W. C. Still, A. Tempczyk, R. C. Hawley, T. Hendrickson, Semianalytical treatment of solvation for molecular mechanics and dynamics, Journal of the American Chemical Society 112 (16) (1990) 6127–6129
work page 1990
-
[62]
A. Onufriev, D. Bashford, D. A.Case, Exploring protein native states and large-scale conformational changes with a modified generalized born model, Proteins: Structure, Function, and Bioinformatics 55 (2) (2004) 383–394
work page 2004
-
[63]
S. W. Lockless, M. Zhou, R. MacKinnon, Structural and thermodynamic properties of selective ion binding in a K+ channel, PLOS BIOLOGY 5 (5) (2007) 1079–1088
work page 2007
-
[64]
J. Tsai, R. Taylor, C. Chothia, M. Gerstein, The packing density in proteins: Standard radii and volumes, Journal of Molecular Biology 290 (1999) 253–266
work page 1999
-
[65]
J. D. Bernal, J. L. Finney, Random close-packed hard-sphere model II. Geometry of random packing of hard spheres, Discussions of the Faraday Society 43 (1967) 62–69
work page 1967
- [66]
-
[67]
Lee, Principles of CAD/CAM/CAE Systems, Addison-Wesley, Boston, 1999
K. Lee, Principles of CAD/CAM/CAE Systems, Addison-Wesley, Boston, 1999. 33 (a) (b) Figure 12: Data structure of REDS and IWDS. (a) REDS, (b) IWDS. 34 Appendix 1. 300 Test PDB Models Table .1: The 300 tested PDB models. 1AA2 1ARL 1BWW 1C26 1CEX 1CT4 1D2K 1D4T 1DC9 1DKL 1DQ0 1DQZ 1E2T 1EAI 1EDQ 1EKG 1EQP 1ES9 1EUM 1EY4 1EZG 1F2V 1F41 1F46 1F60 1FA8 1FCQ 1F...
work page 1999
-
[68]
number of atoms() • Entity locator API (proximity query I) : Twelve APIs
-
[69]
find boundary atoms in van der Waals model()
-
[70]
find buried atoms in van der Waals model()
-
[71]
find first order neighbor atoms in van der Waals model( atom )
-
[72]
find first order neighbor atoms in van der Waals model( atomAr- rangement )
-
[73]
find second order neighbor atoms in van der Waals model( atom )
-
[74]
find second order neighbor atoms in van der Waals model( atom- Arrangement )
-
[75]
find boundary atoms in Lee Richards model( solventProbeRadius )
-
[76]
find buried atoms in Lee Richards model( solventProbeRadius )
-
[77]
find first order neighbor atoms in Lee Richards model( solventProbeRa- dius, atom )
-
[78]
find first order neighbor atoms in Lee Richards model( solventProbeRa- dius, atomArrangement )
-
[79]
find second order neighbor atoms in Lee Richards model( solvent- ProbeRadius, atom )
-
[80]
find second order neighbor atoms in Lee Richards model( solvent- ProbeRadius, atomArrangement ) 36 • Entity verifier API (proximity query II) : Six APIs
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.