Semialgebraic Conditions for Identifying Triangles in Phylogenetic Networks
Pith reviewed 2026-06-26 02:18 UTC · model grok-4.3
The pith
Three Jukes-Cantor network models with embedded triangles produce overlapping but distinct full-dimensional sets of site-pattern probabilities.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
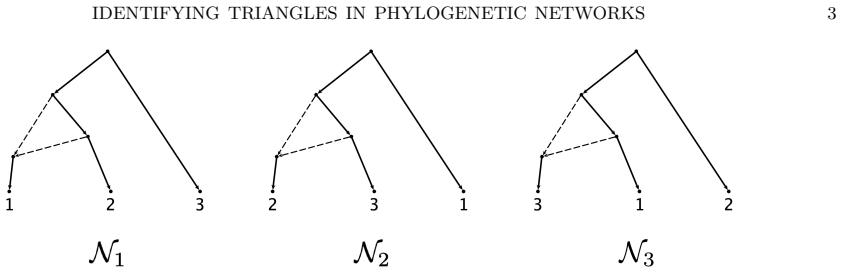
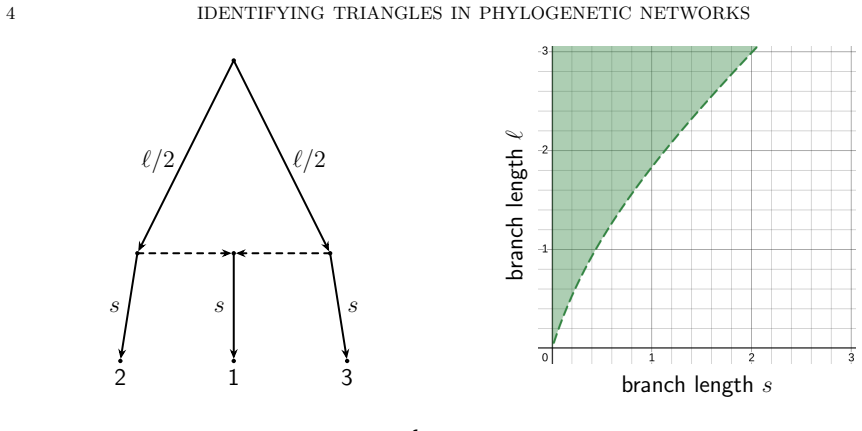
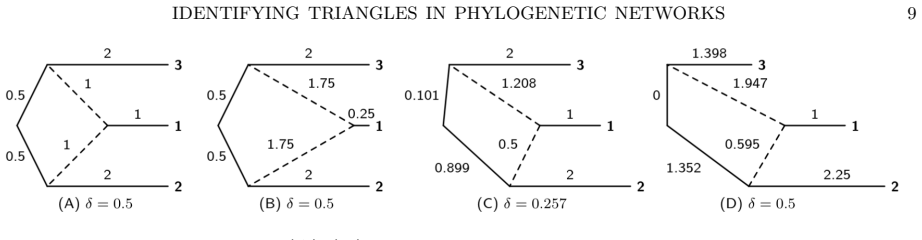
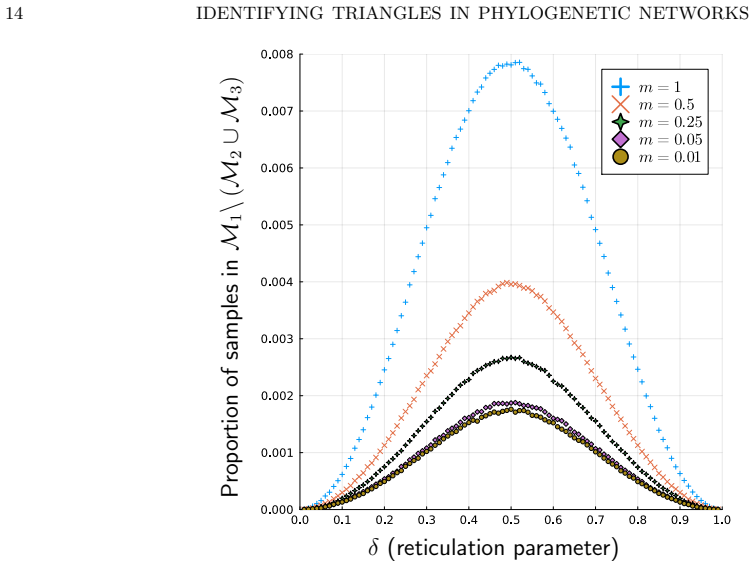
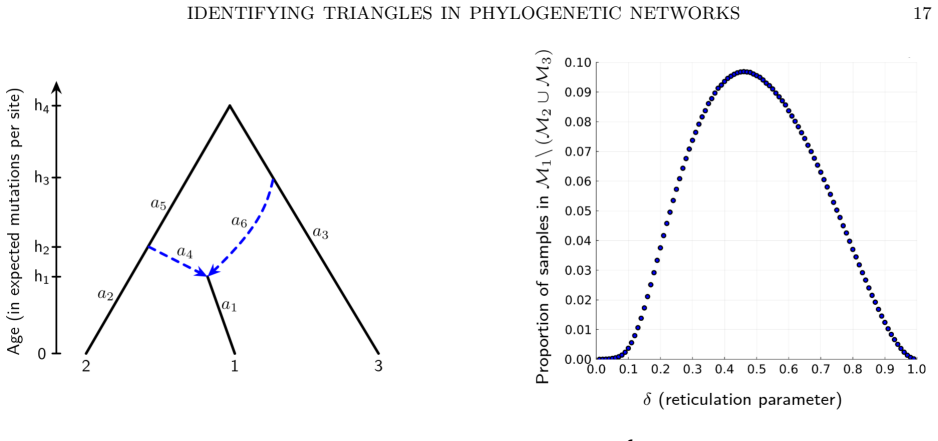
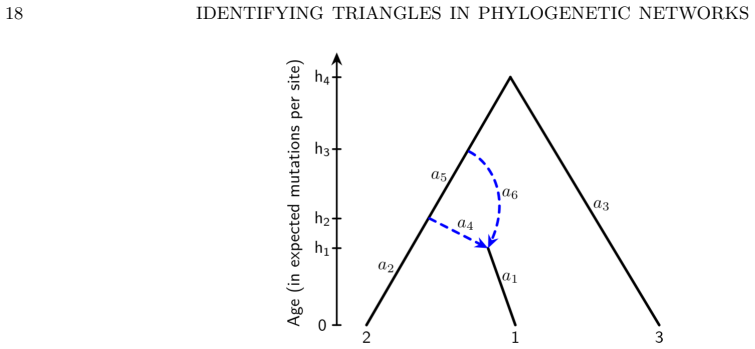
The three 3-leaf Jukes-Cantor phylogenetic network models with embedded triangles admit a complete semialgebraic description. For any pair of these models, both the intersection and the set differences consist of full-dimensional regions in the space of site-pattern probability distributions. Consequently the models are algebraically indistinguishable, not identical, and not identifiable or generically identifiable.
What carries the argument
The semialgebraic sets (regions cut out by polynomial equalities and inequalities) that exactly describe the site-pattern probability distributions for each of the three network models.
Load-bearing premise
Resolving the identifiability question for the three 3-leaf base cases is enough to settle identifiability for arbitrary networks that merely contain embedded triangles.
What would settle it
An explicit probability vector that satisfies the semialgebraic inequalities for exactly one of the three models, or a dimension calculation showing that any intersection or difference has dimension strictly less than the ambient space.
Figures



read the original abstract
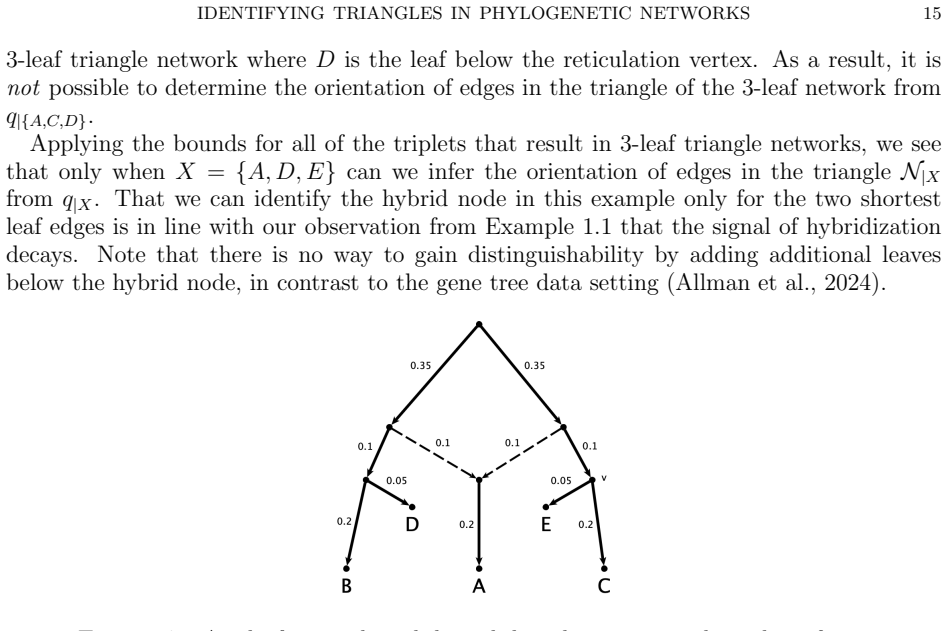
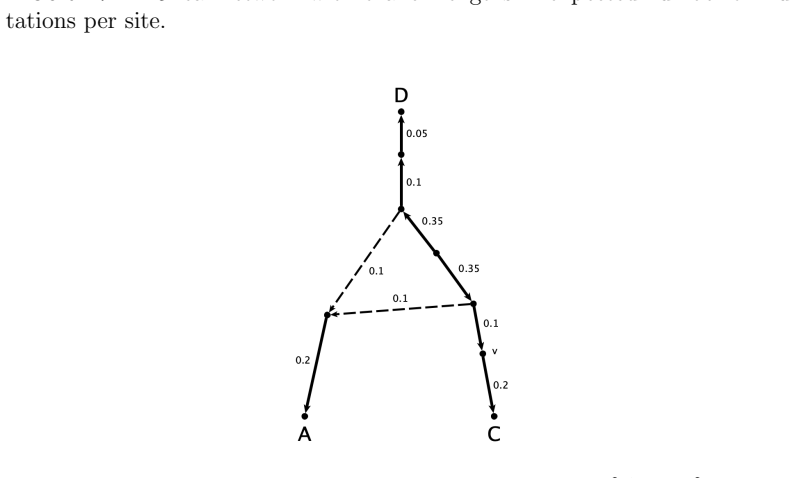
An important consideration for a model-based method of phylogenetic network inference is the identifiability of the network parameter of the model. A recurring theme in previous works exploring this issue is that it is often difficult to identify the orientation of edges in a triangle of the network. In fact, it has been shown that for some models it is impossible to determine the orientation of triangle edges utilizing the standard algebraic technique of phylogenetic invariants. In this work, we consider one such model with a Jukes-Cantor site-substitution process and no coalescence. We give a complete semialgebraic description of three, 3-leaf Jukes-Cantor phylogenetic network models with embedded triangles. By describing these base cases, we resolve several questions about the identifiability of networks with embedded triangles. We show that for any pair of models, the intersection and set differences of the models are full-dimensional regions of the space of site-pattern probability distributions. Thus, despite being algebraically indistinguishable, these network models are not identical, nor are they identifiable (or generically identifiable). Our results also yield a straightforward biological interpretation--that the signal from a hybridization event may be immediately detectable but decays over time until it is impossible to identify the orientation of edges in the triangle of a network.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript computes complete semialgebraic descriptions of three 3-leaf Jukes-Cantor phylogenetic network models with embedded triangles. It shows that pairwise intersections and set differences of these models are full-dimensional subsets of the site-pattern probability simplex. The authors conclude that the models are algebraically indistinguishable yet distinct and non-identifiable (nor generically identifiable), and that the 3-leaf base cases thereby resolve identifiability questions for arbitrary networks containing embedded triangles.
Significance. If the semialgebraic descriptions are correct and the generalization to larger networks is justified, the work supplies a concrete algebraic distinction between models that standard phylogenetic invariants cannot separate. The full-dimensionality results and the biological interpretation of hybridization-signal decay constitute a useful contribution to the literature on network identifiability.
major comments (1)
- [Abstract and final paragraph] Abstract and final paragraph: the claim that describing the three 3-leaf base cases resolves identifiability questions for arbitrary networks with embedded triangles is not accompanied by an explicit reduction argument (e.g., a marginalization lemma, an embedding of the 3-leaf distributions, or a proof that any triangle-orientation ambiguity in a general network projects onto one of the three base cases).
minor comments (1)
- The manuscript would benefit from displaying the explicit polynomials or inequalities that define each semialgebraic set, together with the verification steps used to confirm completeness and full-dimensionality.
Simulated Author's Rebuttal
We thank the referee for their careful reading and constructive feedback on our manuscript. We address the major comment below.
read point-by-point responses
-
Referee: [Abstract and final paragraph] Abstract and final paragraph: the claim that describing the three 3-leaf base cases resolves identifiability questions for arbitrary networks with embedded triangles is not accompanied by an explicit reduction argument (e.g., a marginalization lemma, an embedding of the 3-leaf distributions, or a proof that any triangle-orientation ambiguity in a general network projects onto one of the three base cases).
Authors: We agree that an explicit reduction argument would strengthen the presentation of the generalization. Although the manuscript positions the 3-leaf cases as fundamental base cases (with the implication that marginals on any embedded triangle fall into one of the three models), we acknowledge the absence of a formal statement. In the revised manuscript we will add a short marginalization paragraph establishing that, for any larger network containing an embedded triangle, the induced distribution on those three leaves lies in one of the three semialgebraic sets we describe; the full-dimensionality of the intersections and differences then carries over directly to show that orientation ambiguity persists. This addition clarifies the claim without altering the core semialgebraic results or the 3-leaf theorems. revision: yes
Circularity Check
No circularity; derivation rests on direct semialgebraic computation of 3-leaf base cases
full rationale
The paper computes complete semialgebraic descriptions of three specific 3-leaf Jukes-Cantor network models and verifies that pairwise intersections and symmetric differences are full-dimensional in the probability simplex. These steps are presented as explicit algebraic results rather than reductions to fitted parameters, self-citations, or definitional equivalences. No equations or load-bearing self-citations appear in the abstract or described claims, and the work is self-contained against external algebraic benchmarks. The assertion that 3-leaf cases resolve identifiability for arbitrary networks is an interpretive extension but does not render the central computations tautological by construction.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
- [1]
-
[2]
Allman, E. S., H. Ba \ n os, M. Garrote-Lopez, and J. A. Rhodes. 2024. Identifiability of level-1 species networks from gene tree quartets. Bull. Math. Biol. 86:110. allman2024identifiability
2024
-
[3]
Allman, E. S., H. Ba \ n os, and J. A. Rhodes. 2022. Identifiability of species network topologies from genomic sequences using the logDet distance . J. Math. Biol. 84:35. allman2022identifiability
2022
-
[4]
Ba \ n os, H. 2019. Identifying species network features from gene tree quartets under the coalescent model. Bull. Math. Biol. 81:494--534. banos2019identifying
2019
-
[5]
Barley, A. J., A. Nieto-Montes de Oca, N. L. Manr \' quez-Mor \'a n, and R. C. Thomson. 2022. The evolutionary network of whiptail lizards reveals predictable outcomes of hybridization. Science 377:773--777. barley2022evolutionary
2022
-
[6]
Barnhill, D., M. Garrote-L \'o pez, E. Gross, M. Hill, B. Kagy, J. A. Rhodes, and J. Z. Zhang. 2025. Methodological considerations for semialgebraic hypothesis testing with incomplete U -statistics. arXiv:2507.13531 . barnhill2025methodological
-
[7]
Gross, C
Barton, T., E. Gross, C. Long, and J. Rusinko. 2026. Statistical learning with phylogenetic network invariants. Bull. Soc. Syst. Biol. 4 no. 1 (2026) 4. barton2022statistical
2026
-
[8]
Fernández-Sánchez, and M
Casanellas, M., J. Fernández-Sánchez, and M. Garrote-López. 2021. SAQ: Semi-algebraic quartet reconstruction . IEEE/ACM Trans. Comput. Biol. Bioinform. 18:2855--2861. Casanellas2021SAQ
2021
-
[9]
Schumer, K
Cui, R., M. Schumer, K. Kruesi, R. Walter, P. Andolfatto, and G. G. Rosenthal. 2013. Phylogenomics reveals extensive reticulate evolution in X iphophorus fishes. Evolution 67:2166--2179. Cui2013-ps
2013
-
[10]
Englander, A. K., M. Frohn, E. Gross, N. Holtgrefe, L. van Iersel, M. Jones, and S. Sullivant. 2025. Identifiability of phylogenetic level-2 networks under the Jukes-Cantor model . bioRxiv Pages 2025--04. englander2025identifiability
2025
-
[11]
Evans, S. N. and T. P. Speed. 1993. Invariants of some probability models used in phylogenetic inference. Ann. Stat. Pages 355--377. evans1993invariants
1993
-
[12]
Flouri, T., X. Jiao, B. Rannala, and Z. Yang. 2019. A Bayesian implementation of the multispecies coalescent model with introgression for phylogenomic analysis. Mol. Biol. Evol. 37. Flouri2019
2019
-
[13]
Gambette, P., K. T. Huber, and S. Kelk. 2017. On the challenge of reconstructing level-1 phylogenetic networks from triplets and clusters. J. Math. Biol. 74:1729--1751. Gambette2017-kf
2017
-
[14]
Gross, E. and C. Long. 2018. Distinguishing phylogenetic networks. SIAM J. Appl. Algebra Geom. 2:72--93. gross2018distinguishing
2018
-
[15]
van Iersel, R
Gross, E., L. van Iersel, R. Janssen, M. Jones, C. Long, and Y. Murakami. 2021. Distinguishing level-1 phylogenetic networks on the basis of data generated by Markov processes . J. Math. Biol. 83:1--24. gross2021distinguishing
2021
-
[16]
Hibbins, M. S. and M. W. Hahn. 2022. Phylogenomic approaches to detecting and characterizing introgression. Genetics 220:iyab173. hibbins2022phylogenomic
2022
- [17]
-
[18]
Jukes, T. H., C. R. Cantor, et al. 1969. Evolution of protein molecules. Mammalian protein metabolism 3:132. jukes1969evolution
1969
-
[19]
Garretson, T
Keuler, R., A. Garretson, T. Saunders, R. J. Erickson, N. St. Andre, F. Grewe, H. Smith, H. T. Lumbsch, J.-P. Huang, L. L. St. Clair, and S. D. Leavitt. 2020. Genome-scale data reveal the role of hybridization in lichen-forming fungi. Sci. Rep. 10:1497. keuler2020genome
2020
-
[20]
Krantz, S. and H. Parks. 2008. Geometric integration theory. Springer. krantz2008geometric
2008
-
[21]
Langdon, Q. K., J. S. Groh, S. M. Aguillon, D. L. Powell, T. Gunn, C. Payne, J. J. Baczenas, A. Donny, T. O. Dodge, K. Du, et al. 2024. Swordtail fish hybrids reveal that genome evolution is surprisingly predictable after initial hybridization. PLoS Biology 22:e3002742. langdon2024swordtail
2024
-
[22]
Leavitt, S. D., F. Fern \'a ndez-Mendoza, S. P \'e rez-Ortega, M. Sohrabi, P. K. Divakar, J. Vondr \'a k, H. Thorsten Lumbsch, and L. L. S. Clair. 2013. Local representation of global diversity in a cosmopolitan lichen-forming fungal species complex ( Rhizoplaca , Ascomycota ). J. Biogeogr. 40:1792--1806. leavitt2013local
2013
-
[23]
Mallet, J. 2005. Hybridization as an invasion of the genome. Trends Ecol. Evol. 20:229--237. mallet2005hybridization
2005
-
[24]
Beltr \'a n, W
Mallet, J., M. Beltr \'a n, W. Neukirchen, and M. Linares. 2007. Natural hybridization in heliconiine butterflies: the species boundary as a continuum. BMC Evol. Biol. 7:28. mallet2007natural
2007
-
[25]
Besansky, and M
Mallet, J., N. Besansky, and M. W. Hahn. 2016. How reticulated are species? BioEssays 38:140--149. mallet2016reticulated
2016
-
[26]
Holtgrefe, V
Martin, S., N. Holtgrefe, V. Moulton, and R. M. Leggett. 2025. Algebraic invariants for inferring 4-leaf semi-directed phylogenetic networks. Syst. Biol. Page syaf071. martin2023algebraic
2025
-
[27]
Moran, B. M., C. Payne, Q. Langdon, D. L. Powell, Y. Brandvain, and M. Schumer. 2021. The genomic consequences of hybridization. Elife 10:e69016. moran2021genomic
2021
-
[28]
Pardi, F. and C. Scornavacca. 2015. Reconstructible phylogenetic networks: do not distinguish the indistinguishable. PLoS Comput. Biol. 11:e1004135. Pardi2015-ix
2015
-
[29]
Pe \ n alba, J. V., A. Runemark, J. I. Meier, P. Singh, G. O. Wogan, R. S \'a nchez-Guill \'e n, J. Mallet, S. J. Rometsch, M. Menon, O. Seehausen, et al. 2024. The role of hybridization in species formation and persistence. Cold Spring Harb. Perspect. Biol. 16:a041445. penalba2024role
2024
-
[30]
Rhodes, J. A., H. Ba \ n os, J. Xu, and C. An \'e . 2025. Identifying circular orders for blobs in phylogenetic networks. Adv. Appl. Math. 163:102804. rhodes2025identifying
2025
-
[31]
Rose, J. P., B. Li, M. J. Sporck-Koehler, E. A. Stacy, K. R. Wood, E. M. Lemmon, A. R. Lemmon, C. Ané, K. J. Sytsma, and T. J. Givnish. 2025. Phylogenomics of the tetraploid Hawaiian lobeliads: Implications for their origin, dispersal history, and adaptive radiation. Proc. Natl. Acad. Sci. U.S.A. 122:e2421004122. lobeloids
2025
-
[32]
Steel, et al
Semple, C., M. Steel, et al. 2003. Phylogenetics vol. 24. Oxford University Press. semple2003phylogenetics
2003
-
[33]
Solís-Lemus, C. and C. Ané. 2016. Inferring phylogenetic networks with maximum pseudolikelihood under incomplete lineage sorting. PLoS Genet. 12:e1005896. solislemus2016snaq
2016
-
[34]
Drton, and D
Sturma, N., M. Drton, and D. Leung. 2024. Testing many constraints in possibly irregular models using incomplete U -statistics. J. R. Stat. Soc. Ser. B Stat Methodol. 86:987--1012. SturmaNilsDrton
2024
-
[35]
Sturmfels, B. and S. Sullivant. 2005. Toric ideals of phylogenetic invariants. J. Comput. Biol. 12:457--481. SS05
2005
-
[36]
Sullivant, S. 2023. Algebraic Statistics vol. 194. AMS. sullivant2023algebraic
2023
-
[37]
The algebraic-phylogenetics collaboration . 2026. A database of small trees and networks in algebraic phylogenetics. Version 0.3. Available at http://www.algebraicphylogenetics.org. smalltrees
2026
-
[38]
van der Heijden, E. S. M., K. Näsvall, F. A. Seixas, C. E. B. Nobre, A. C. D. Maia, P. Salazar-Carrión, J. M. Walker, D. Szczerbowski, S. Schulz, I. A. Warren, K. G. G. Córdova, M. J. Sánchez-Carvajal, F. Chandi, A. P. Arias-Cruz, N. Rueda-M, C. Salazar, K. K. Dasmahapatra, S. H. Montgomery, M. McClure, D. E. Absolon, T. C. Mathers, C. A. Santos, S. McCar...
2025
-
[39]
Veller, C., N. B. Edelman, P. Muralidhar, and M. A. Nowak. 2023. Recombination and selection against introgressed DNA . Evolution 77:1131--1144. veller2023recombination
2023
-
[40]
Wen, D., Y. Yu, J. Zhu, and L. Nakhleh. 2018. Inferring phylogenetic networks using PhyloNet . Syst. Biol. 67:735--740. phylonet-paper
2018
-
[41]
Xu, J. and C. An \'e . 2023. Identifiability of local and global features of phylogenetic networks from average distances. J. Math. Biol. 86:12. xu2023identifiability
2023
-
[42]
Zhang, C., H. A. Ogilvie, A. J. Drummond, and T. Stadler. 2018. Bayesian inference of species networks from multilocus sequence data. Mol. Biol. Evol. 35:504--517. Zhang2018-zk
2018
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.