SF-Cluster: Frustration-Guided MSA Subsampling for Alternative Protein Conformation Recovery
Pith reviewed 2026-07-02 00:39 UTC · model grok-4.3
The pith
SF-Cluster subsamples MSAs via local energetic frustration patterns to recover alternative protein conformations more reliably than sequence-space methods.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
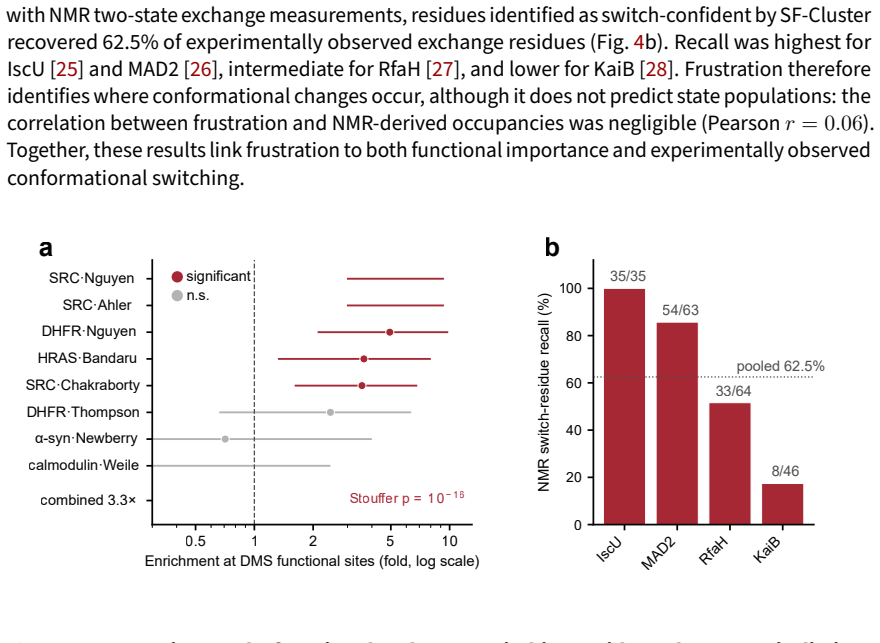
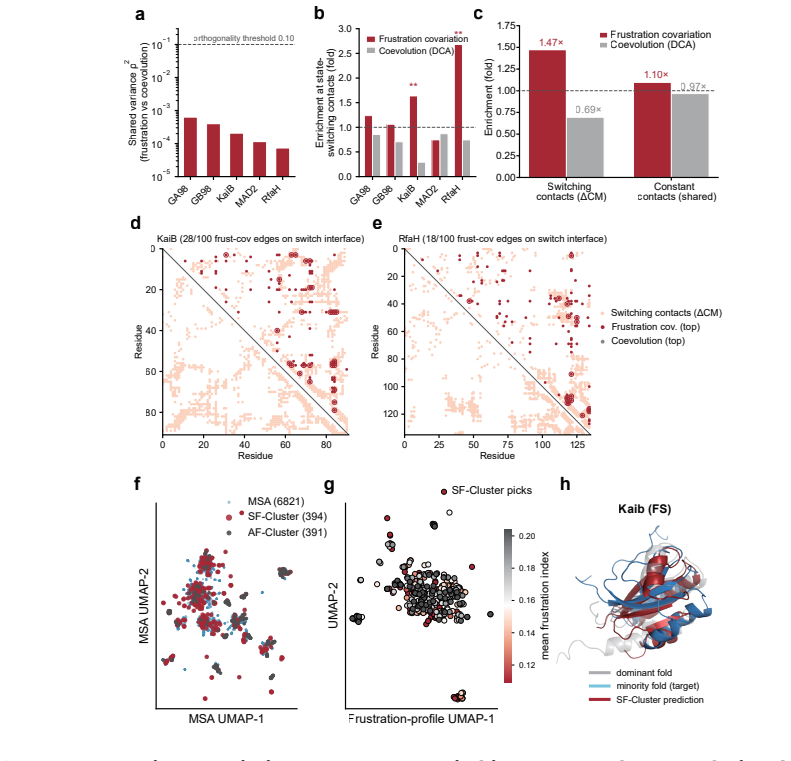
SF-Cluster improves target-state recovery of the alternative conformation over AF-Cluster across the two-state classes in a benchmark of 48 cases, with the largest improvement observed for allosteric systems. The recovery advantage is largely explained by the effective depth of the selected subsets, which frustration-pattern selection reliably reaches. At the same time, highly frustrated residues are enriched at sites supported by deep mutational scanning and NMR two-state exchange, and frustration covariation is enriched at state-switching contacts while remaining distinct from coevolutionary coupling.
What carries the argument
Frustration-pattern-based MSA subsampling, in which predicted local energetic frustration patterns serve as the guide for choosing alignment subsets that favor one conformational basin over another.
If this is right
- The selected MSAs transfer to an architecturally distinct predictor, showing that the conformational signal resides in MSA composition.
- Matched-depth controls indicate that the recovery advantage is largely explained by the effective depth reached by frustration-pattern selection.
- Highly frustrated residues are enriched at sites supported by deep mutational scanning and NMR two-state exchange.
- Frustration covariation is enriched at state-switching contacts yet remains distinct from coevolutionary coupling.
Where Pith is reading between the lines
- The same frustration signal could be tested as an input feature for predictors that do not rely on MSAs at all.
- Frustration-guided selection might be combined with other orthogonal signals such as predicted contacts or evolutionary couplings to further refine basin targeting.
- The enrichment of frustration at experimentally validated switching sites suggests a route to prioritize residues for mutagenesis experiments aimed at shifting conformational equilibria.
Load-bearing premise
That patterns of predicted local energetic frustration form a signal largely independent of sequence similarity and that this signal reliably encodes which conformational basin an MSA subset will favor.
What would settle it
A new benchmark of two-state proteins in which frustration-selected MSAs produce no recovery gain over AF-Cluster once MSA depth is matched, or in which the selected alignments fail to transfer to a second predictor.
Figures




read the original abstract
Deep-learning structure predictors are sensitive to their multiple sequence alignment (MSA) input, making MSA subsampling a practical route to recovering alternative conformations. Existing approaches such as AF-Cluster operate in sequence space, providing limited control over which conformational basin is sampled. We introduce SF-Cluster, which subsamples MSAs using patterns of predicted local energetic frustration, a representation largely independent of sequence similarity. Across a benchmark of 48 cases spanning fold-switching, allosteric, oligomerization-coupled, and intrinsically disordered systems, and using an AF-Cluster-style dual-reference RMSD criterion, SF-Cluster improves target-state recovery of the alternative conformation over AF-Cluster across the two-state classes, with the largest improvement observed for allosteric systems (+15.5 percentage points). The selected MSAs transfer to an architecturally distinct predictor, indicating that the conformational signal resides in MSA composition. Mechanistically, matched-depth controls show that this recovery advantage is largely explained by the effective depth of the selected subsets, which frustration-pattern selection reliably reaches. At the same time, highly frustrated residues are enriched at sites supported by deep mutational scanning and NMR two-state exchange, and frustration covariation is enriched at state-switching contacts while remaining distinct from coevolutionary coupling. Together, these results identify frustration patterns as a transferable representation for conformational prediction and position MSA subsampling as a representation-guided reweighting problem.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces SF-Cluster, a method for subsampling MSAs using patterns of predicted local energetic frustration to recover alternative protein conformations with deep-learning predictors. On a benchmark of 48 cases across fold-switching, allosteric, oligomerization, and disordered systems, and using an AF-Cluster-style dual-reference RMSD criterion, SF-Cluster improves target-state recovery over AF-Cluster (largest gain +15.5 pp in allosteric systems). The selected MSAs transfer to an architecturally distinct predictor; frustration is enriched at DMS- and NMR-supported sites and at state-switching contacts (distinct from coevolution); matched-depth controls indicate that recovery gains are largely explained by the effective depth reached by frustration-pattern selection.
Significance. If the benchmark results, transferability, and orthogonal validations hold, the work supplies a practical MSA-subsampling tool, empirical evidence that effective depth is a dominant factor in conformational recovery, and biological support linking local frustration to two-state dynamics. It frames MSA subsampling as representation-guided reweighting and identifies frustration patterns as a transferable signal, which could guide future methods even if the primary mechanism is depth selection.
major comments (1)
- [Abstract] Abstract: The claim that frustration patterns supply 'a representation largely independent of sequence similarity' that 'captures conformational basin information' is in tension with the statement that 'matched-depth controls show that this recovery advantage is largely explained by the effective depth of the selected subsets.' If depth-matched random subsampling yields statistically indistinguishable recovery rates, the method reduces to reliable depth reweighting rather than frustration-guided basin targeting; the manuscript should directly compare SF-Cluster to depth-matched random controls and revise the mechanistic interpretation accordingly.
minor comments (2)
- [Abstract] Abstract: No error bars, confidence intervals, or statistical significance tests are reported for the percentage-point improvements or enrichment statistics.
- [Abstract] Abstract: The frustration prediction method is referenced but not described at even a high level (e.g., which energy function or software is used), making it impossible to assess reproducibility from the abstract alone.
Simulated Author's Rebuttal
We thank the referee for their careful and constructive review. We address the single major comment below.
read point-by-point responses
-
Referee: [Abstract] Abstract: The claim that frustration patterns supply 'a representation largely independent of sequence similarity' that 'captures conformational basin information' is in tension with the statement that 'matched-depth controls show that this recovery advantage is largely explained by the effective depth of the selected subsets.' If depth-matched random subsampling yields statistically indistinguishable recovery rates, the method reduces to reliable depth reweighting rather than frustration-guided basin targeting; the manuscript should directly compare SF-Cluster to depth-matched random controls and revise the mechanistic interpretation accordingly.
Authors: We acknowledge the tension in the abstract wording. The manuscript already reports matched-depth controls showing that recovery gains are largely explained by the effective depth reliably reached via frustration-pattern selection. This supports interpreting the method primarily as representation-guided depth reweighting rather than direct basin targeting. We agree the phrase 'captures conformational basin information' risks overstating the case and will revise the abstract to align the mechanistic claims with the depth-based findings while retaining the statements on independence from sequence similarity (supported by the method design) and the orthogonal enrichment results at DMS-, NMR-, and state-switching sites. The matched-depth controls already provide the requested comparison to depth-matched selection; we will ensure this is stated explicitly in the revised text. revision: yes
Circularity Check
No significant circularity; claims rest on external benchmarks and depth-matched controls
full rationale
The paper evaluates on an external 48-case benchmark using an AF-Cluster-style dual-reference RMSD criterion and explicitly reports matched-depth controls showing that recovery gains are largely attributable to effective MSA depth reached by the selection procedure. No equations, fitted parameters, or self-citations are shown that reduce the reported improvements or the frustration representation claim to quantities defined by the authors' own prior fits or inputs. The central positioning of frustration patterns as a transferable signal is supported by enrichment observations (deep mutational scanning, NMR, state-switching contacts) that remain distinct from coevolutionary coupling, keeping the derivation self-contained against external data.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Extant fold-switching proteins are widespread.Proceed- ings of the National Academy of Sciences, 115(23):5968–5973, 2018
Lauren L Porter and Loren L Looger. Extant fold-switching proteins are widespread.Proceed- ings of the National Academy of Sciences, 115(23):5968–5973, 2018
2018
-
[2]
Functional and regulatory roles of fold-switching proteins
Allen K Kim and Lauren L Porter. Functional and regulatory roles of fold-switching proteins. Structure, 29(1):6–14, 2021
2021
-
[3]
TD3B: Transition-Directed Discrete Diffusion for Allosteric Binder Gener- ation
Hanqun Cao, Aastha Pal, Sophia Tang, Yinuo Zhang, Jingjie Zhang, Pheng-Ann Heng, and Pranam Chatterjee. TD3B: Transition-Directed Discrete Diffusion for Allosteric Binder Gener- ation. InForty-third International Conference on Machine Learning (Spotlight), 2026
2026
-
[4]
Highly accurate protein structure prediction with alphafold.nature, 596(7873):583–589, 2021
John Jumper, Richard Evans, Alexander Pritzel, Tim Green, Michael Figurnov, Olaf Ron- neberger, Kathryn Tunyasuvunakool, Russ Bates, Augustin Žídek, Anna Potapenko, et al. Highly accurate protein structure prediction with alphafold.nature, 596(7873):583–589, 2021
2021
-
[5]
Alphafold2 fails to predict protein fold switching
Devlina Chakravarty and Lauren L Porter. Alphafold2 fails to predict protein fold switching. Protein Science, 31(6):e4353, 2022
2022
-
[6]
Alphafold predictions of fold-switched conformations are driven by structure memorization.Nature communications, 15(1):7296, 2024
Devlina Chakravarty, Joseph W Schafer, Ethan A Chen, Joseph F Thole, Leslie A Ronish, Myeongsang Lee, and Lauren L Porter. Alphafold predictions of fold-switched conformations are driven by structure memorization.Nature communications, 15(1):7296, 2024
2024
-
[7]
Sampling alternative conformational states of transporters and receptors with alphafold2.elife, 11:e75751, 2022
Diego Del Alamo, Davide Sala, Hassane S Mchaourab, and Jens Meiler. Sampling alternative conformational states of transporters and receptors with alphafold2.elife, 11:e75751, 2022
2022
-
[8]
Speach_af: Sampling protein ensembles and conformational heterogeneity with alphafold2.PLoS computational biology, 18(8):e1010483, 2022
Richard A Stein and Hassane S Mchaourab. Speach_af: Sampling protein ensembles and conformational heterogeneity with alphafold2.PLoS computational biology, 18(8):e1010483, 2022
2022
-
[9]
Predicting multiple conformations via sequence clustering and alphafold2.Nature, 625(7996):832–839, 2024
Hannah K Wayment-Steele, Adedolapo Ojoawo, Renee Otten, Julia M Apitz, Warintra Pit- sawong, Marc Hömberger, Sergey Ovchinnikov, Lucy Colwell, and Dorothee Kern. Predicting multiple conformations via sequence clustering and alphafold2.Nature, 625(7996):832–839, 2024
2024
-
[10]
Leveraging sequence purification for accurate prediction of multiple conformational states with alphafold2.Research Square, pages rs–3, 2025
Enming Xing, Junjie Zhang, Shen Wang, and Xiaolin Cheng. Leveraging sequence purification for accurate prediction of multiple conformational states with alphafold2.Research Square, pages rs–3, 2025
2025
-
[11]
Structure prediction of alternative protein conformations
Patrick Bryant and Frank Noé. Structure prediction of alternative protein conformations. Nature Communications, 15(1):7328, 2024. 15
2024
-
[12]
Large-scale predictions of alternative protein conformations by alphafold2-based sequence association.Nature Communications, 16(1):5622, 2025
Myeongsang Lee, Joseph W Schafer, Jeshuwin Prabakaran, Devlina Chakravarty, Madeleine F Clore, and Lauren L Porter. Large-scale predictions of alternative protein conformations by alphafold2-based sequence association.Nature Communications, 16(1):5622, 2025
2025
-
[13]
Disentangling coevolutionary constraints for modeling protein conformational heterogeneity.Communica- tions Chemistry, 9:146, 2026
Shimian Li, Chengwei Zhang, Lupeng Kong, Yue Xue, Sirui Liu, and Yi Qin Gao. Disentangling coevolutionary constraints for modeling protein conformational heterogeneity.Communica- tions Chemistry, 9:146, 2026
2026
-
[14]
Frustrai-seq: Scaling local energetic frustration to the protein sequence space.bioRxiv, pages 2026–02, 2026
Jan-Philipp Leusch, Miriam Poley-Gil, Miguel Fernandez-Martin, Nicola Bordin, Burkhard Rost, R Gonzalo Parra, and Michael Heinzinger. Frustrai-seq: Scaling local energetic frustration to the protein sequence space.bioRxiv, pages 2026–02, 2026
2026
-
[15]
Frustration in biomolecules
Diego U Ferreiro, Elizabeth A Komives, and Peter G Wolynes. Frustration in biomolecules. Quarterly reviews of biophysics, 47(4):285–363, 2014
2014
-
[16]
On the role of frustration in the energy landscapes of allosteric proteins.Proceedings of the National Academy of Sciences, 108(9):3499–3503, 2011
Diego U Ferreiro, Joseph A Hegler, Elizabeth A Komives, and Peter G Wolynes. On the role of frustration in the energy landscapes of allosteric proteins.Proceedings of the National Academy of Sciences, 108(9):3499–3503, 2011
2011
-
[17]
Cath–a hierarchic classification of protein domain structures.Structure, 5(8): 1093–1109, 1997
Christine A Orengo, Alex D Michie, Susan Jones, David T Jones, Mark B Swindells, and Janet M Thornton. Cath–a hierarchic classification of protein domain structures.Structure, 5(8): 1093–1109, 1997
1997
-
[18]
Boltz-1 democratizing biomolecular interaction modeling.BioRxiv, pages 2024–11, 2025
Jeremy Wohlwend, Gabriele Corso, Saro Passaro, Noah Getz, Mateo Reveiz, Ken Leidal, Wojtek Swiderski, Liam Atkinson, Tally Portnoi, Itamar Chinn, et al. Boltz-1 democratizing biomolecular interaction modeling.BioRxiv, pages 2024–11, 2025
2024
-
[19]
Proteingym: Large- scale benchmarks for protein fitness prediction and design.Advances in neural information processing systems, 36:64331–64379, 2023
Pascal Notin, Aaron Kollasch, Daniel Ritter, Lood Van Niekerk, Steffanie Paul, Han Spinner, Nathan Rollins, Ada Shaw, Rose Orenbuch, Ruben Weitzman, et al. Proteingym: Large- scale benchmarks for protein fitness prediction and design.Advances in neural information processing systems, 36:64331–64379, 2023
2023
-
[20]
A combined approach reveals a regulatory mechanism coupling src’s kinase activity, localization, and phosphotransferase-independent functions.Molecular cell, 74(2):393–408, 2019
Ethan Ahler, Ames C Register, Sujata Chakraborty, Linglan Fang, Emily M Dieter, Katherine A Sitko, Rama Subba Rao Vidadala, Bridget M Trevillian, Martin Golkowski, Hannah Gelman, et al. A combined approach reveals a regulatory mechanism coupling src’s kinase activity, localization, and phosphotransferase-independent functions.Molecular cell, 74(2):393–408, 2019
2019
-
[21]
Molecular determinants of hsp90 dependence of src kinase revealed by deep mutational scanning.Protein Science, 32(7):e4656, 2023
Vanessa Nguyen, Ethan Ahler, Katherine A Sitko, Jason J Stephany, Dustin J Maly, and Dou- glas M Fowler. Molecular determinants of hsp90 dependence of src kinase revealed by deep mutational scanning.Protein Science, 32(7):e4656, 2023
2023
-
[22]
Deconstruction of the ras switching cycle through saturation mutagenesis.Elife, 6: e27810, 2017
Pradeep Bandaru, Neel H Shah, Moitrayee Bhattacharyya, John P Barton, Yasushi Kondo, Joshua C Cofsky, Christine L Gee, Arup K Chakraborty, Tanja Kortemme, Rama Ranganathan, et al. Deconstruction of the ras switching cycle through saturation mutagenesis.Elife, 6: e27810, 2017
2017
-
[23]
A framework for exhaustively mapping functional missense variants.Molecular systems biology, 13(12):MSB177908, 2017
Jochen Weile, Song Sun, Atina G Cote, Jennifer Knapp, Marta Verby, Joseph C Mellor, Yingzhou Wu, Carles Pons, Cassandra Wong, Natascha van Lieshout, et al. A framework for exhaustively mapping functional missense variants.Molecular systems biology, 13(12):MSB177908, 2017. 16
2017
-
[24]
Deep mutational scanning reveals the structural basis forα-synuclein activity.Nature chemical biology, 16(6):653–659, 2020
Robert W Newberry, Jaime T Leong, Eric D Chow, Martin Kampmann, and William F De- Grado. Deep mutational scanning reveals the structural basis forα-synuclein activity.Nature chemical biology, 16(6):653–659, 2020
2020
-
[25]
Metamorphic protein iscu changes conformation by cis–trans isomerizations of two peptidyl–prolyl peptide bonds.Biochemistry, 51(48): 9595–9602, 2012
Ziqi Dai, Marco Tonelli, and John L Markley. Metamorphic protein iscu changes conformation by cis–trans isomerizations of two peptidyl–prolyl peptide bonds.Biochemistry, 51(48): 9595–9602, 2012
2012
-
[26]
The mad2 spindle checkpoint protein has two distinct natively folded states.Nature structural & molecular biology, 11(4):338–345, 2004
Xuelian Luo, Zhanyun Tang, Guohong Xia, Katja Wassmann, Tomohiro Matsumoto, Josep Rizo, and Hongtao Yu. The mad2 spindle checkpoint protein has two distinct natively folded states.Nature structural & molecular biology, 11(4):338–345, 2004
2004
-
[27]
An α helix to β barrel domain switch transforms the transcription factor rfah into a translation factor.Cell, 150(2):291–303, 2012
Björn M Burmann, Stefan H Knauer, Anastasia Sevostyanova, Kristian Schweimer, Rachel A Mooney, Robert Landick, Irina Artsimovitch, and Paul Rösch. An α helix to β barrel domain switch transforms the transcription factor rfah into a translation factor.Cell, 150(2):291–303, 2012
2012
-
[28]
A protein fold switch joins the circadian oscillator to clock output in cyanobacteria.Science, 349(6245):324–328, 2015
Yong-Gang Chang, Susan E Cohen, Connie Phong, William K Myers, Yong-Ick Kim, Roger Tseng, Jenny Lin, Li Zhang, Joseph S Boyd, Yvonne Lee, et al. A protein fold switch joins the circadian oscillator to clock output in cyanobacteria.Science, 349(6245):324–328, 2015
2015
-
[29]
Unsupervisedly prompting alphafold2 for accurate few-shot protein structure prediction.Journal of Chemical Theory and Computation, 19(22): 8460–8471, 2023
Jun Zhang, Sirui Liu, Mengyun Chen, Haotian Chu, Min Wang, Zidong Wang, Jialiang Yu, Ningxi Ni, Fan Yu, Dechin Chen, et al. Unsupervisedly prompting alphafold2 for accurate few-shot protein structure prediction.Journal of Chemical Theory and Computation, 19(22): 8460–8471, 2023
2023
-
[30]
Msa generation with seqs2seqs pre- training: advancing protein structure predictions.Advances in Neural Information Processing Systems, 37:57324–57348, 2024
Le Zhang, Jiayang Chen, Tao Shen, Yu Li, and Siqi Sun. Msa generation with seqs2seqs pre- training: advancing protein structure predictions.Advances in Neural Information Processing Systems, 37:57324–57348, 2024
2024
-
[31]
Msagpt: Neural prompting protein structure prediction via msa generative pre-training.Advances in Neural Information Processing Systems, 37:37504–37534, 2024
Bo Chen, Zhilei Bei, Xingyi Cheng, Pan Li, Jie Tang, and Le Song. Msagpt: Neural prompting protein structure prediction via msa generative pre-training.Advances in Neural Information Processing Systems, 37:37504–37534, 2024
2024
-
[32]
Plame: Lightweight msa design advances protein folding from evolu- tionary embeddings
Hanqun Cao, Xinyi Zhou, Zijun Gao, Chenyu Wang, Xin Gao, Zhi Zhang, Chunbin Gu, Ge Liu, and Pheng-Ann Heng. Plame: Lightweight msa design advances protein folding from evolu- tionary embeddings. InNeurIPS 2025 AI for Science Workshop, 2025
2025
-
[33]
Generative power of a protein language model trained on multiple sequence alignments.Elife, 12:e79854, 2023
Damiano Sgarbossa, Umberto Lupo, and Anne-Florence Bitbol. Generative power of a protein language model trained on multiple sequence alignments.Elife, 12:e79854, 2023
2023
-
[34]
Msaflow: a unified approach for msa representation, augmentation, and family-based protein design
Anirudh Venkatraman, Hanqun Cao, Tong Wei, Chaoran Cheng, and Ge Liu. Msaflow: a unified approach for msa representation, augmentation, and family-based protein design. InNeurIPS 2025 AI for Science Workshop
2025
-
[35]
Localizing frustration in native proteins and protein assemblies.Proceedings of the National Academy of Sciences, 104(50):19819–19824, 2007
Diego U Ferreiro, Joseph A Hegler, Elizabeth A Komives, and Peter G Wolynes. Localizing frustration in native proteins and protein assemblies.Proceedings of the National Academy of Sciences, 104(50):19819–19824, 2007
2007
-
[36]
17 Local frustration around enzyme active sites.Proceedings of the National Academy of Sciences, 116(10):4037–4043, 2019
Maria I Freiberger, A Brenda Guzovsky, Peter G Wolynes, R Gonzalo Parra, and Diego U Ferreiro. 17 Local frustration around enzyme active sites.Proceedings of the National Academy of Sciences, 116(10):4037–4043, 2019
2019
-
[37]
Colabfold: making protein folding accessible to all.Nature methods, 19 (6):679–682, 2022
Milot Mirdita, Konstantin Schütze, Yoshitaka Moriwaki, Lim Heo, Sergey Ovchinnikov, and Martin Steinegger. Colabfold: making protein folding accessible to all.Nature methods, 19 (6):679–682, 2022
2022
-
[38]
Tm-align: a protein structure alignment algorithm based on the tm-score.Nucleic acids research, 33(7):2302–2309, 2005
Yang Zhang and Jeffrey Skolnick. Tm-align: a protein structure alignment algorithm based on the tm-score.Nucleic acids research, 33(7):2302–2309, 2005
2005
-
[39]
Protein frustratometer: a tool to localize energetic frustration in protein molecules.Nucleic acids research, 40(W1):W348–W351, 2012
Michael Jenik, R Gonzalo Parra, Leandro G Radusky, Adrian Turjanski, Peter G Wolynes, and Diego U Ferreiro. Protein frustratometer: a tool to localize energetic frustration in protein molecules.Nucleic acids research, 40(W1):W348–W351, 2012
2012
-
[40]
Protein frustratometer 2: a tool to localize energetic frustration in protein molecules, now with electrostatics.Nucleic acids research, 44(W1): W356–W360, 2016
R Gonzalo Parra, Nicholas P Schafer, Leandro G Radusky, Min-Yeh Tsai, A Brenda Guzovsky, Peter G Wolynes, and Diego U Ferreiro. Protein frustratometer 2: a tool to localize energetic frustration in protein molecules, now with electrostatics.Nucleic acids research, 44(W1): W356–W360, 2016. 18 Supplementary Information S1 Implementation Details S1.1 Matched...
2016
-
[41]
ThenP(B s | A ′ 1)̸=P(B s | A ′ 2)
̸= ϕ(A′ 2), and that for some basinB s and at least oner̸=s, gs(ϕ(A′ 1))−g r(ϕ(A′ 1)) ̸= gs(ϕ(A′ 2))−g r(ϕ(A′ 2)) . ThenP(B s | A ′ 1)̸=P(B s | A ′ 2). Proof.The log-odds between basinssandrunder any sub-MSAA ′ is log P(B s | A ′) P(B r | A ′) =g s(ϕ(A′))−g r(ϕ(A′)). By assumption this quantity differs betweenA′ 1 and A′ 2 for at least one r̸ = s, so the ...
-
[42]
̸= P (Bs | A ′ 2). Theorem S2.1 establishes that MSA subsampling is a conformational reweighting operation whose effect is mediated entirely by the shift in representationϕ(A′), making the choice of ϕ the binding constraint on what subsampling can achieve. 31 S2.3 Theorem 2: Conditions for focusing and impossibility Theorem S2.2.(a) Targeted focusing.Supp...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.