DiSTILL: A Hybrid Cloud-HPC Workflow System for Reproducible Spatial Transcriptomics Analysis
Pith reviewed 2026-07-01 06:22 UTC · model grok-4.3
The pith
DiSTILL generates run-specific execution bundles and SLURM scripts from cloud registries to make spatial transcriptomics analyses reproducible on hybrid HPC setups.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
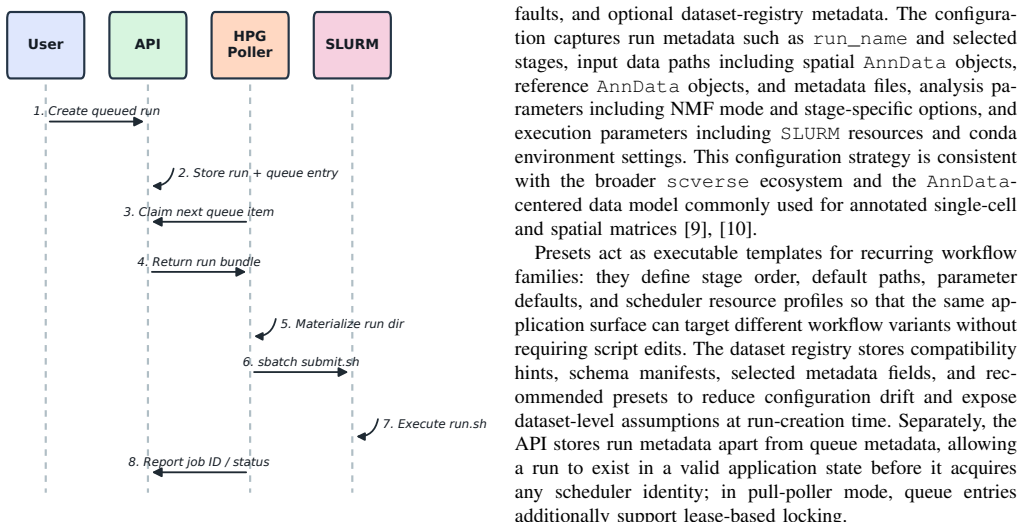
DiSTILL is a hybrid cloud-HPC workflow system that uses an API backend built with FastAPI, a web frontend, a dataset and preset registry, and a Python pipeline generator to materialize run-specific execution bundles and SLURM submission scripts, supporting local, SSH-mediated, and pull-based poller execution modes for reproducible spatial transcriptomics analysis.
What carries the argument
The Python pipeline generator that materializes run-specific execution bundles and SLURM submission scripts from dataset and preset registries.
If this is right
- Analyses gain queue-based orchestration and auditable artifacts without manual script editing.
- HPC submission becomes possible in environments that restrict persistent API automation.
- The same presets and datasets can be reused across multiple execution modes.
- Workflows wrap existing pipelines into an application layer that handles configuration semantics.
Where Pith is reading between the lines
- The registry could support versioned sharing of analysis presets between separate research teams.
- The bundle generation step might reduce errors when scaling from small test runs to full HPC jobs.
- Similar hybrid designs could apply to other notebook-heavy bioinformatics pipelines that require HPC resources.
Load-bearing premise
User-supplied datasets must satisfy the schema assumptions of the wrapped analytical pipeline.
What would settle it
Submit a dataset that violates the pipeline schema and check whether the generated bundles produce consistent or invalid outputs.
Figures




read the original abstract
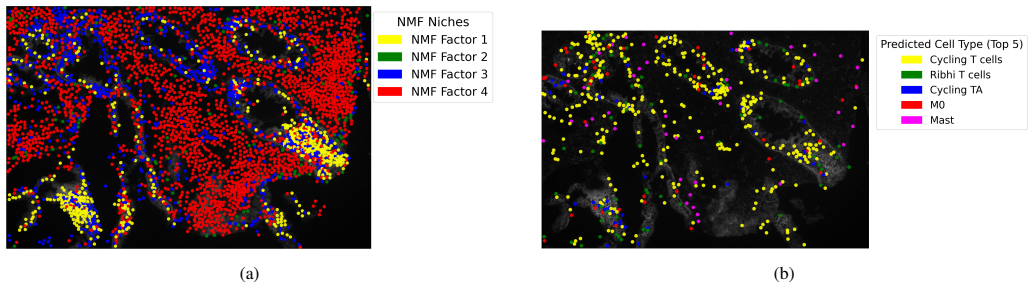
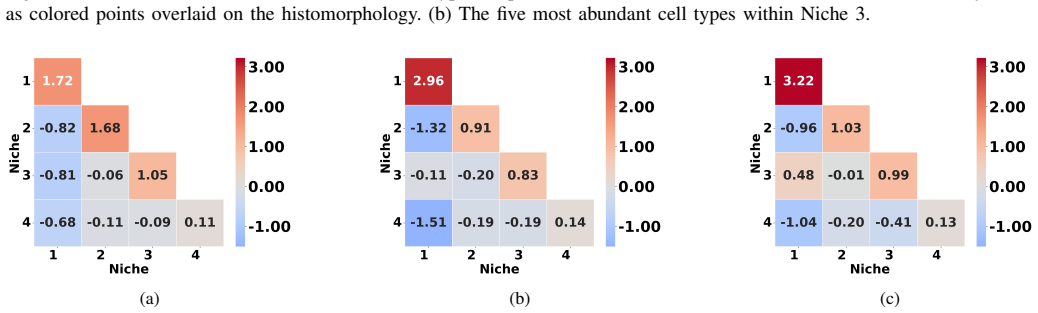
Spatial transcriptomics workflows increasingly combine large annotated data objects, notebook-based analyses, and resource-intensive statistical models that must be executed on high-performance computing (HPC) systems. In practice, these workflows are often difficult to reproduce because configuration, validation, stage execution, and artifact handling are fragmented across $\textit{ad hoc}$ scripts and manually edited notebooks. We present $\textit{DiSTILL}$ (Disease Diagnosis from Spatial Transcriptomics via Interpretable Latent Learning), a hybrid cloud$-$HPC workflow system for reproducible spatial transcriptomics (ST) analysis. DiSTILL combines an application programming interface (API) backend built with $\texttt{FastAPI}$, a web frontend, a dataset and preset registry, and a Python pipeline generator that materializes run-specific execution bundles and $\texttt{SLURM}$ submission scripts. The system supports local, Secure Shell (SSH)-mediated, and pull-based poller execution modes, enabling HPC submission in environments where persistent API-initiated automation is restricted. We describe the system through the lens of an inflammatory bowel disease (IBD) ST workflow that operationalizes the analytical pipeline of Tan $\textit{et al.}$ into an auditable application layer. Accordingly, the contribution of this paper is a workflow systems contribution centered on reproducible execution, queue-based orchestration, configuration semantics, and deployment across a split cloud$-$HPC architecture. The broader application goal of DiSTILL is to support user-supplied datasets that satisfy the schema assumptions of the wrapped analytical pipeline.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents DiSTILL, a hybrid cloud-HPC workflow system for reproducible spatial transcriptomics analysis. It integrates a FastAPI API backend, web frontend, dataset and preset registry, and Python pipeline generator that materializes run-specific execution bundles and SLURM submission scripts. The system supports local, SSH-mediated, and pull-based poller execution modes. It is illustrated via an IBD ST workflow that wraps the analytical pipeline of Tan et al., with the contribution framed as a workflow systems paper on reproducible execution, queue-based orchestration, configuration semantics, and split cloud-HPC deployment. The scope is limited to user-supplied datasets satisfying the wrapped pipeline's schema assumptions.
Significance. If implemented and shown to function as described, DiSTILL could reduce fragmentation in ST workflows by supplying an auditable layer for configuration, validation, stage execution, and artifact handling across cloud and HPC resources. The explicit scoping to schema-compliant datasets and the three execution modes are internally consistent with the reproducibility goals. However, the manuscript supplies no implementation details, validation results, benchmarks, or error-handling evidence, so the practical significance and reliability remain unevaluated.
major comments (1)
- [Abstract] Abstract: the central claim that DiSTILL constitutes a reproducible workflow system is unsupported by any implementation details, validation results, benchmarks, or error-handling evidence, leaving the reproducibility and deployment claims without demonstrated support.
Simulated Author's Rebuttal
We thank the referee for the detailed review and constructive feedback. We address the single major comment below and outline planned revisions to strengthen the manuscript.
read point-by-point responses
-
Referee: [Abstract] Abstract: the central claim that DiSTILL constitutes a reproducible workflow system is unsupported by any implementation details, validation results, benchmarks, or error-handling evidence, leaving the reproducibility and deployment claims without demonstrated support.
Authors: We agree that the abstract and current manuscript text emphasize architectural claims without accompanying empirical validation or benchmarks. The contribution is framed as a workflow-systems description centered on configuration semantics, queue-based orchestration, and split cloud-HPC deployment rather than performance evaluation; the IBD pipeline serves only as an illustrative wrapper. To address the concern, we will revise the abstract to qualify the reproducibility claims as design-level guarantees and add a dedicated Implementation and Error Handling section that includes pseudocode for the pipeline generator, registry validation logic, and the three execution modes, plus a brief discussion of failure modes observed during development. We will also make the source repository publicly available with the revised submission. No runtime benchmarks are planned, as they fall outside the stated scope. revision: yes
Circularity Check
No significant circularity detected
full rationale
The paper is a descriptive systems contribution detailing the architecture of DiSTILL (FastAPI backend, web frontend, dataset registry, Python pipeline generator, and three execution modes) for wrapping an existing analytical pipeline. No equations, fitted parameters, predictions, or derivation chains appear in the text. The scoping statement that DiSTILL supports datasets satisfying the wrapped pipeline's schema assumptions is an explicit boundary condition rather than a hidden dependency. Self-citation to Tan et al. is present but functions only as an external reference to the wrapped pipeline and is not load-bearing for the reproducibility or deployment claims of the workflow layer itself. The argument is therefore self-contained as a software description with no reduction of outputs to inputs by construction.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption User-supplied datasets satisfy the schema assumptions of the wrapped analytical pipeline.
Reference graph
Works this paper leans on
-
[1]
M. J. T. Tan, M. Kapetanaki, and P. V . Benos, “Engineering spatial and molecular features from cellular niches to inform predictions of inflammatory bowel disease,” Sep. 2025, arXiv:2509.09923, revised Jan. 2026
-
[2]
Computational approaches and challenges in spatial transcriptomics,
S. Fang, B. Chen, Y . Zhang, H. Sun, L. Liuet al., “Computational approaches and challenges in spatial transcriptomics,”Genomics, Pro- teomics & Bioinformatics, vol. 21, no. 1, pp. 24–47, 2023
work page 2023
-
[3]
Computational methods for alignment and inte- gration of spatially resolved transcriptomics data,
Y . Liu and C. Yang, “Computational methods for alignment and inte- gration of spatially resolved transcriptomics data,”Computational and Structural Biotechnology Journal, vol. 23, pp. 1094–1105, 2024
work page 2024
-
[4]
Cell-type deconvolution methods for spatial transcriptomics,
L. C. Gaspard-Boulinc, L. Gortana, T. Walter, E. Barillot, and F. M. G. Cavalli, “Cell-type deconvolution methods for spatial transcriptomics,” Nature Reviews Genetics, vol. 26, pp. 828–846, 2025
work page 2025
-
[5]
Practical computational reproducibility in the life sciences,
B. Gr ¨uning, J. Chilton, J. K ¨oster, and R. Dale, “Practical computational reproducibility in the life sciences,”Cell Systems, vol. 6, no. 6, pp. 631–635, 2018
work page 2018
-
[6]
Provenance information for biomedical data and workflows: Scoping review,
K. Gierend, F. Kr ¨uger, S. Genehr, F. Hartmann, F. Siegel, D. Waltemath, T. Ganslandt, and A. A. Zeleke, “Provenance information for biomedical data and workflows: Scoping review,”Journal of Medical Internet Research, vol. 26, p. e51297, 2024
work page 2024
-
[7]
Applying the fair principles to computational workflows,
S. R. Wilkinson, M. Aloqalaa, K. Belhajjame, M. R. Crusoe, B. d. P. Kinoshita, L. Gadelha, D. Garijoet al., “Applying the fair principles to computational workflows,”Scientific Data, vol. 12, p. 328, 2025
work page 2025
-
[8]
SCANPY: large-scale single- cell gene expression data analysis,
F. A. Wolf, P. Angerer, and F. J. Theis, “SCANPY: large-scale single- cell gene expression data analysis,”Genome Biology, vol. 19, no. 1, p. 15, 2018
work page 2018
-
[9]
The scverse project provides a computational ecosystem for single-cell omics data analysis,
I. Virshup, S. Rybakov, F. J. Theis, P. Angerer, F. A. Wolfet al., “The scverse project provides a computational ecosystem for single-cell omics data analysis,”Nature Biotechnology, vol. 41, pp. 604–606, 2023
work page 2023
-
[10]
anndata: Access and store annotated data matrices,
I. Virshup, S. Rybakov, F. J. Theis, P. Angerer, and F. A. Wolf, “anndata: Access and store annotated data matrices,”Journal of Open Source Software, vol. 9, no. 101, p. 4371, 2024
work page 2024
-
[11]
Cell2location maps fine-grained cell types in spatial transcriptomics,
V . Kleshchevnikov, A. Shmatko, E. Dann, A. Aivazidis, H. W. King, T. Li, R. Elmentaite, A. Lomakin, V . Kedlian, A. Gayoso, M. S. Jain, J. S. Park, L. Ramona, L. Mazutis, F. A. Wolf, F. J. Theis, L. K. James, T. D. Hether, T. J. Stevens, K. Saeb-Parsy, O. A. Bayraktar, C. Yapp, and S. A. Teichmann, “Cell2location maps fine-grained cell types in spatial t...
work page 2022
-
[12]
Giotto: a toolbox for integrative analysis and visualization of spatial expression data,
R. Dries, Q. Zhu, R. Dong, C.-H. L. Eng, H. Li, K. Liu, Y . Fu, T. Zhao, A. Sarkar, F. Bao, R. E. George, E. Pierson, L. Cai, and G.-C. Yuan, “Giotto: a toolbox for integrative analysis and visualization of spatial expression data,”Genome Biology, vol. 22, p. 78, 2021
work page 2021
-
[13]
Squidpy: a scalable framework for spatial omics analysis,
G. Palla, H. Spitzer, M. Klein, D. Fischer, A. C. Schaar, L. B. Kuem- merle, S. Rybakov, I. L. Ibarra, O. Holmberg, I. Virshup, M. Lotfollahi, S. Richter, F. J. Theis, P. V . Kharchenko, M. Hemberg, D. Pe’er, and F. A. Wolf, “Squidpy: a scalable framework for spatial omics analysis,” Nature Methods, vol. 19, pp. 171–178, 2022
work page 2022
-
[14]
Information-theoretic methods in spatial transcriptomics,
V . G. Hinostroza Fuentes, M. J. T. Tan, M. Kapetanaki, P. Rashidi, K. Huang, and P. V . Benos, “Information-theoretic methods in spatial transcriptomics,” Feb. 2026, techRxiv preprint, doi:10.36227/techrxiv.177155947.75111980/v1
-
[15]
Snakemake—a scalable bioinformatics workflow engine,
J. K ¨oster and S. Rahmann, “Snakemake—a scalable bioinformatics workflow engine,”Bioinformatics, vol. 28, no. 19, pp. 2520–2522, 2012
work page 2012
-
[16]
Nextflow enables reproducible computational work- flows,
P. Di Tommaso, M. Chatzou, E. W. Floden, P. P. Barja, E. Palumbo, and C. Notredame, “Nextflow enables reproducible computational work- flows,”Nature Biotechnology, vol. 35, no. 4, pp. 316–319, 2017
work page 2017
-
[17]
The galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update,
E. Afganet al., “The galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update,”Nucleic Acids Research, vol. 46, no. W1, pp. W537–W544, 2018
work page 2018
-
[18]
Panpipes: a pipeline for multiomic single-cell and spatial transcriptomic data analysis,
F. Curionet al., “Panpipes: a pipeline for multiomic single-cell and spatial transcriptomic data analysis,”Genome Biology, vol. 25, p. 181, 2024
work page 2024
-
[19]
Spatialone: end-to-end analysis of visium data at scale,
M. Kamelet al., “Spatialone: end-to-end analysis of visium data at scale,”Bioinformatics, vol. 40, no. 9, p. btae509, 2024
work page 2024
-
[20]
SLURM: Simple linux utility for resource management,
A. B. Yoo, M. A. Jette, and M. Grondona, “SLURM: Simple linux utility for resource management,” inJob Scheduling Strategies for Parallel Processing, ser. Lecture Notes in Computer Science, 2003, vol. 2862, pp. 44–60
work page 2003
-
[21]
T. C. Lovelace, M. Dudek, J. Fiore, and P. V . Benos,rCausalMGM: Scalable Causal Discovery and Model Selection on Mixed Datasets with ’rCausalMGM’, 2026, r package version 1.0. [Online]. Available: https://CRAN.R-project.org/package=rCausalMGM
work page 2026
-
[22]
A unified approach to interpreting model predictions,
S. M. Lundberg and S.-I. Lee, “A unified approach to interpreting model predictions,” inAdvances in Neural Information Processing Systems, 2017
work page 2017
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.