BEAGLE 4.1: A high-performance library for computation on phylogenetic trees across diverse parallel architectures
Pith reviewed 2026-06-29 00:25 UTC · model grok-4.3
The pith
BEAGLE 4.1 adds gradient algorithms and hardware support that deliver up to fourfold CPU and tenfold model speedups on phylogenetic likelihood calculations.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
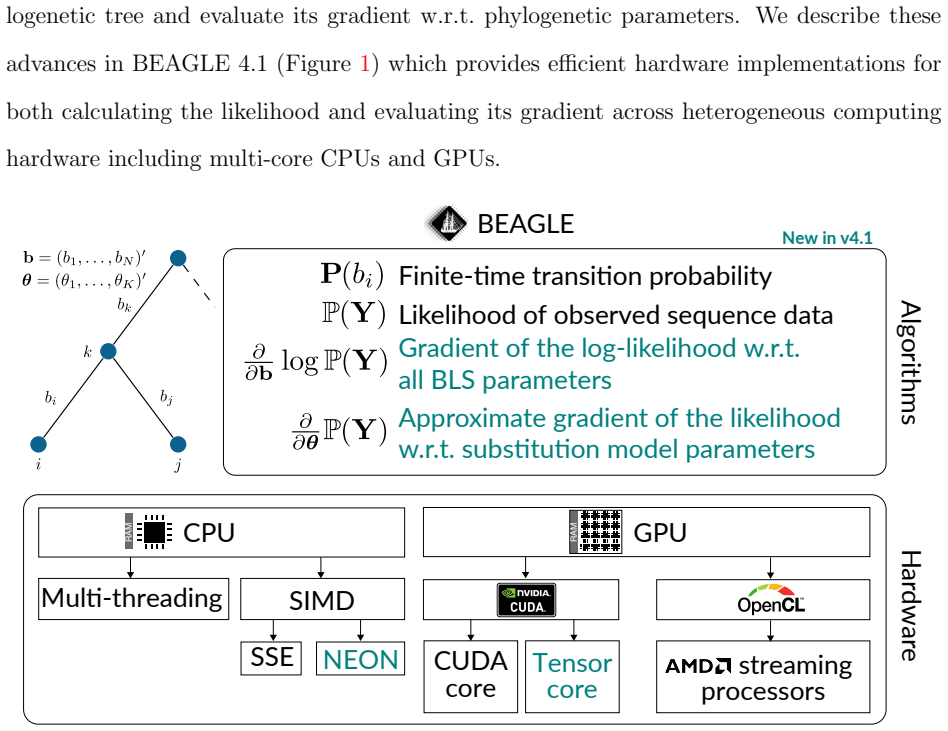
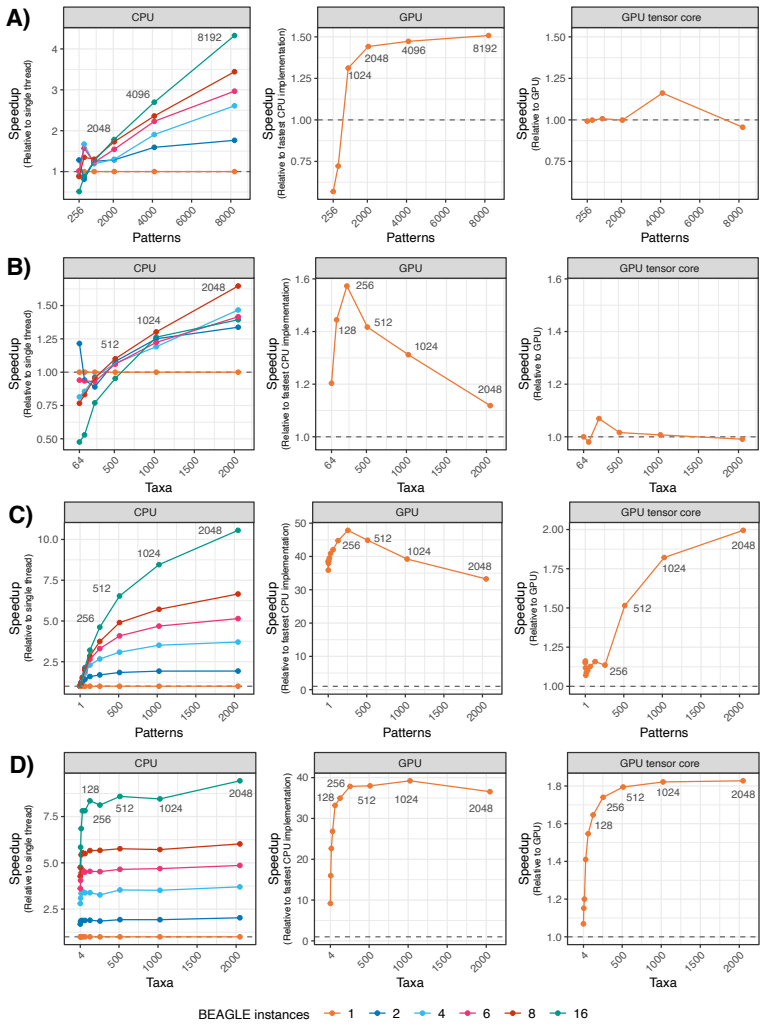
We present BEAGLE 4.1, which incorporates new algorithms to evaluate gradients on phylogenetic trees and provides new hardware implementations for both likelihoods and gradients supporting ARM NEON intrinsics and optimized matrix multiplication units called tensor cores on NVIDIA GPUs. We benchmark the performance scaling of the library across a number of patterns and taxa on multi-core CPUs and GPUs, and compare the speedup afforded by NVIDIA and AMD GPUs as well as performance scaling with an increasing number of GPUs. We show that multi-core CPU implementations provide up to a fourfold speedup over single-threaded CPU implementations and up to a tenfold speedup for nucleotide and codon mo
What carries the argument
New algorithms for evaluating sequence likelihoods and their high-dimensional gradients on phylogenetic trees, together with hardware-specific implementations for multi-core CPUs, ARM NEON, and GPU tensor cores.
If this is right
- Phylogenetic software packages can integrate the new gradient algorithms and hardware code to accelerate both maximum-likelihood and Bayesian inference.
- Performance gains increase with larger numbers of taxa and site patterns, favoring analyses of bigger trees.
- Codon models receive substantially higher GPU speedups than nucleotide models even when the number of taxa or patterns is small.
- Tensor cores on NVIDIA GPUs supply an extra twofold acceleration specifically for codon models relative to standard CUDA cores.
- ARM CPUs obtain up to 1.3-fold improvement from NEON instructions, though the gain shrinks when eight or more threads are used.
Where Pith is reading between the lines
- Packages that already call BEAGLE for likelihoods can obtain gradient-based methods such as Hamiltonian Monte Carlo without writing new low-level code.
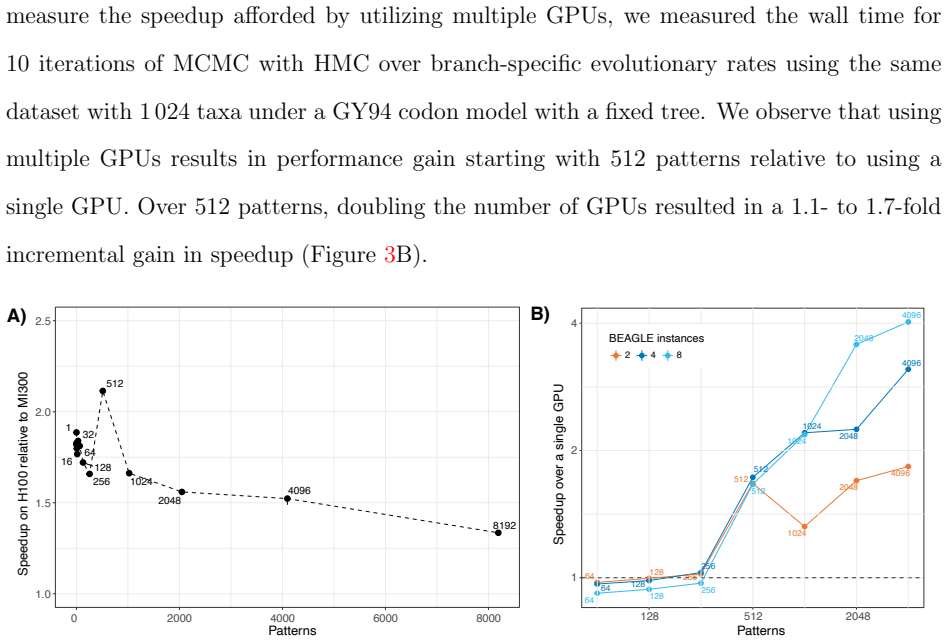
- The scaling behavior suggests that GPU clusters become attractive for routine codon-model analyses that were previously limited to small trees.
- ARM-based servers or laptops could become viable platforms for moderate-sized phylogenetic work once NEON support is used.
- If gradient calculations dominate runtime in many current tools, the reported factors imply that overall inference wall time could drop enough to allow more extensive model comparison on the same hardware.
Load-bearing premise
The speedups measured on selected patterns and taxa numbers will hold for the full range of real phylogenetic datasets and inference tasks that users run after integrating the library.
What would settle it
Measure wall-clock time for likelihood-plus-gradient evaluation on a real dataset containing thousands of taxa and thousands of site patterns using a production phylogenetic package integrated with the library; the central claim fails if the observed factor is below twofold on multi-core CPUs or shows no gain on GPUs for codon models.
Figures
read the original abstract
Efficient evaluation of sequence data likelihoods and their high-dimensional gradients on phylogenetic trees improves inference under both maximum-likelihood and Bayesian frameworks. Here, we present BEAGLE 4.1, a high-performance library for statistical phylogenetics that incorporates new algorithms to evaluate these gradients on phylogenetic trees. We also provide new hardware implementations for both likelihoods and gradients supporting ARM NEON intrinsics and optimized matrix multiplication units -- called tensor cores -- on NVIDIA graphics processing units (GPUs). We benchmark the performance scaling of the library across a number of patterns and taxa on multi-core CPUs and GPUs, and compare the speedup afforded by NVIDIA and AMD GPUs as well as performance scaling with an increasing number of GPUs. We show that multi-core CPU implementations provide up to a fourfold speedup over single-threaded CPU implementations and up to an tenfold speedup for nucleotide and codon models, respectively, with performance generally improving as the number of taxa and site patterns increases. GPUs outperform multi-threaded CPU implementations for a realistic number of patterns, even for nucleotide models with a small state-space size of 4, while for codon models they provide substantially higher performance gains even for a single pattern or four taxa. Tensor cores on GPUs provide up to 2-fold speedup relative to standard CUDA cores for codon models. Using NEON instructions on ARM CPUs affords up to a $\sim 1.3$-fold speedup over non-SIMD implementation with the speedup going down to 1.1-fold at 8 CPU threads. We provide these new algorithms to evaluate the gradient and efficient hardware implementations for both likelihood and gradient calculations through BEAGLE 4.1, such that they can be readily integrated into phylogenetic software packages.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents BEAGLE 4.1, an updated high-performance library for evaluating phylogenetic likelihoods and their gradients on trees. It introduces new algorithms for gradient computation, hardware-specific implementations including ARM NEON intrinsics on CPUs and tensor-core matrix multiplication on NVIDIA GPUs, and reports benchmarks across patterns and taxa on multi-core CPUs, GPUs from multiple vendors, and multi-GPU setups. The central performance claims are up to 4-fold speedup on multi-core CPUs for nucleotide models and 10-fold for codon models relative to single-threaded, with GPUs providing additional gains that increase with taxa and site patterns, and tensor cores yielding up to 2-fold improvement for codon models.
Significance. If the benchmark results hold under realistic conditions, the work provides substantial practical value by supplying optimized, integrable code for both likelihood and gradient calculations that can accelerate maximum-likelihood and Bayesian phylogenetic inference across common hardware platforms. The explicit support for gradients, ARM architectures, and tensor cores addresses current needs in large-scale analyses.
major comments (3)
- [Benchmarking section] Benchmarking section: The performance scaling results are described only as having been obtained 'across a number of patterns and taxa,' without listing the concrete values of taxa counts, site-pattern counts, substitution models, or tree sizes used in the timing experiments. This information is required to assess whether the reported 4×/10× speedups and the statement that 'performance generally improving as the number of taxa and site patterns increases' generalize beyond the tested regime.
- [Gradient evaluation and timing] Gradient evaluation and timing: The manuscript introduces new algorithms for gradient computation but does not state whether the reported CPU and GPU timing runs included these gradient calculations or were restricted to likelihood-only evaluations. Because the abstract and introduction emphasize gradients as a key motivation for the library update, the absence of this detail directly affects the strength of the utility claims for modern inference workflows.
- [GPU vs. CPU comparison] GPU vs. CPU comparison: The claim that 'GPUs outperform multi-threaded CPU implementations for a realistic number of patterns' is not accompanied by a definition or citation for what constitutes a realistic pattern count, nor by any measurement of host-package integration overhead. Without these, the practical speedup for end-user phylogenetic software cannot be evaluated.
minor comments (1)
- [Abstract] Abstract: 'up to an tenfold speedup' contains a grammatical error and should read 'up to a tenfold speedup.'
Simulated Author's Rebuttal
We thank the referee for their constructive comments. We address each major comment below and indicate planned revisions to the manuscript.
read point-by-point responses
-
Referee: [Benchmarking section] Benchmarking section: The performance scaling results are described only as having been obtained 'across a number of patterns and taxa,' without listing the concrete values of taxa counts, site-pattern counts, substitution models, or tree sizes used in the timing experiments. This information is required to assess whether the reported 4×/10× speedups and the statement that 'performance generally improving as the number of taxa and site patterns increases' generalize beyond the tested regime.
Authors: We agree that listing the concrete values is necessary for assessing generalizability. The revised manuscript will include a table or subsection specifying the taxa counts, site-pattern counts, substitution models, and tree sizes used in the timing experiments. revision: yes
-
Referee: [Gradient evaluation and timing] Gradient evaluation and timing: The manuscript introduces new algorithms for gradient computation but does not state whether the reported CPU and GPU timing runs included these gradient calculations or were restricted to likelihood-only evaluations. Because the abstract and introduction emphasize gradients as a key motivation for the library update, the absence of this detail directly affects the strength of the utility claims for modern inference workflows.
Authors: The reported timing runs included the gradient calculations. We will add an explicit statement in the benchmarking section of the revised manuscript clarifying this to strengthen the utility claims. revision: yes
-
Referee: [GPU vs. CPU comparison] GPU vs. CPU comparison: The claim that 'GPUs outperform multi-threaded CPU implementations for a realistic number of patterns' is not accompanied by a definition or citation for what constitutes a realistic pattern count, nor by any measurement of host-package integration overhead. Without these, the practical speedup for end-user phylogenetic software cannot be evaluated.
Authors: We will revise the text to define realistic pattern counts with supporting citations from the phylogenetic literature. The reported benchmarks measure core library performance; we will add a note that host-package integration overhead is typically negligible based on prior BEAGLE work, while acknowledging it was not directly measured here. revision: partial
Circularity Check
No circularity: implementation and benchmark paper with no derivations or fitted models
full rationale
The paper describes a software library (BEAGLE 4.1) and reports empirical timing measurements on CPUs and GPUs for likelihood and gradient evaluations. No mathematical derivations, parameter fitting, or predictive models are presented whose outputs could reduce to their inputs by construction. The central claims rest on direct benchmarks rather than any self-referential logic, self-citation chains, or ansatzes. This matches the expected non-circular case for an engineering/performance contribution.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Systematic Biology , author =. 2019 , pages =. doi:10.1093/sysbio/syz020 , abstract =
-
[2]
Nonparametric Modeling of Continuous-Time Markov Chains
Nonparametric Modeling of Continuous-Time Markov Chains , author=. arXiv preprint arXiv:2511.03954 , year=
work page internal anchor Pith review Pith/arXiv arXiv
-
[3]
Molecular Biology and Evolution , author =
Gradients. Molecular Biology and Evolution , author =. 2020 , pages =. doi:10.1093/molbev/msaa130 , abstract =
-
[4]
Systematic Biology , author =. 2012 , pages =. doi:10.1093/sysbio/syr100 , language =
-
[5]
CUDA Programming: A Developer’s Guide to Parallel Computing with GPUs , year =
Cook, Shane , edition =. CUDA Programming: A Developer’s Guide to Parallel Computing with GPUs , year =
-
[6]
and Gohara, David and Shi, Guochun , doi =
Stone, John E. and Gohara, David and Shi, Guochun , doi =. Computing in Science & Engineering , langid =
-
[7]
Journal of Molecular Evolution , author =
Evolutionary trees from. Journal of Molecular Evolution , author =. 1981 , keywords =. doi:10.1007/BF01734359 , abstract =
-
[8]
, date-added =
Neal, Radford M. , date-added =. Handbook of Markov Chain Monte Carlo , number =
-
[9]
Numerical methods for unconstrained optimization and nonlinear equations , Volume =
Dennis Jr, John E and Schnabel, Robert B , Publisher =. Numerical methods for unconstrained optimization and nonlinear equations , Volume =
-
[10]
Random-. Systematic Biology , author =. 2024 , pages =. doi:10.1093/sysbio/syae019 , language =
-
[11]
2020 , howpublished =
NVIDIA , title =. 2020 , howpublished =
2020
-
[12]
Scalable. Systematic Biology , author =. 2023 , pages =. doi:10.1093/sysbio/syad039 , abstract =
-
[13]
Gangavarapu, Karthik and Ji, Xiang and Shao, Yucai and Rambaut, Andrew and Lemey, Philippe and Baele, Guy and Suchard, Marc A , title =. Systematic Biology , pages =. 2026 , month =. doi:10.1093/sysbio/syag017 , url =
-
[14]
Journal of Molecular Evolution , author =
Dating of the human-ape splitting by a molecular clock of mitochondrial. Journal of Molecular Evolution , author =. 1985 , keywords =. doi:10.1007/BF02101694 , abstract =
-
[15]
Molecular Biology and Evolution , volume =
Goldman, N and Yang, Z , title =. Molecular Biology and Evolution , volume =. 1994 , month =. doi:10.1093/oxfordjournals.molbev.a040153 , url =
-
[16]
Tavar. Am. Math. Soc , volume=
-
[17]
and Brusselmans, Marius and Dudas, Gytis and Ji, Xiang and McCrone, John T
Baele, Guy and Carvalho, Luiz M. and Brusselmans, Marius and Dudas, Gytis and Ji, Xiang and McCrone, John T. and Lemey, Philippe and Suchard, Marc A. and Rambaut, Andrew , title =. 2024 , doi =. https://www.biorxiv.org/content/early/2024/12/10/2024.12.08.627395.full.pdf , journal =
2024
-
[18]
sse2neon: Translating Intel SSE intrinsics to Arm NEON , year =
-
[19]
B ayesian phylogenetic and phylodynamic data integration using BEAST 1.10
Suchard, Marc A and Lemey, Philippe and Baele, Guy and Ayres, Daniel L and Drummond, Alexei J and Rambaut, Andrew. B ayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol
-
[20]
PLOS Computational Biology , publisher =. 2019 , month =. doi:10.1371/journal.pcbi.1006650 , author =
-
[21]
and Darling, Aaron and Höhna, Sebastian and Larget, Bret and Liu, Liang and Suchard, Marc A
Ronquist, Fredrik and Teslenko, Maxim and van der Mark, Paul and Ayres, Daniel L. and Darling, Aaron and Höhna, Sebastian and Larget, Bret and Liu, Liang and Suchard, Marc A. and Huelsenbeck, John P. , title =. Systematic Biology , volume =. 2012 , month =. doi:10.1093/sysbio/sys029 , url =
-
[22]
Nature Methods , author =. 2025 , keywords =. doi:10.1038/s41592-025-02751-x , language =
-
[23]
Guindon, Stéphane and Dufayard, Jean-François and Lefort, Vincent and Anisimova, Maria and Hordijk, Wim and Gascuel, Olivier , title =. Systematic Biology , volume =. 2010 , month =. doi:10.1093/sysbio/syq010 , url =
-
[24]
Smith, Killian and Ayres, Daniel and Neumaier, René and Wörheide, Gert and Höhna, Sebastian , title =. Systematic Biology , volume =. 2024 , month =. doi:10.1093/sysbio/syae005 , url =
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.