Molecular Docking via Weighted Subgraph Isomorphism on Quantum Annealers
Pith reviewed 2026-05-24 02:27 UTC · model grok-4.3
The pith
Molecular docking is cast as weighted subgraph isomorphism between a ligand graph and a protein pocket grid, then solved as QUBO on quantum annealers.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The authors formulate molecular docking as a weighted subgraph isomorphism problem between the ligand graph (encoding geometry and flexibility) and the weighted spatial grid of the protein pocket, encode the resulting matching task as a QUBO, solve it on quantum annealers, and compare the outcomes with those obtained from classical simulated annealing on the same instances.
What carries the argument
Weighted subgraph isomorphism between the ligand graph and the pocket grid, mapped to a QUBO for annealing solvers.
If this is right
- Quantum annealers become applicable to the pose-search phase of docking without intermediate classical preprocessing steps.
- Ligand flexibility is handled directly by the graph representation rather than by enumerating discrete conformers.
- Energetic preferences of the pocket are incorporated through edge weights in the grid, allowing the QUBO to reflect binding affinity contributions.
- Performance can be benchmarked on identical QUBO instances against classical simulated annealing to quantify any quantum advantage.
Where Pith is reading between the lines
- If hardware noise and embedding overhead decrease, the same graph formulation could be applied to larger pockets or more flexible ligands than current classical exhaustive searches allow.
- The approach could be combined with downstream classical scoring functions that refine the ranked poses returned by the annealer.
- Similar graph-isomorphism encodings might be tested on other geometry-constrained problems in computational chemistry, such as protein-protein interface prediction.
Load-bearing premise
The ligand graph and the weighted pocket grid together contain all the geometric and energetic information required to recover correct binding poses.
What would settle it
On a ligand-protein pair with a known crystal structure, the lowest-energy configuration returned by the annealer lies more than 2 Å RMSD from the experimental pose.
Figures













read the original abstract
Molecular docking is an essential step in the drug discovery process involving the detection of three-dimensional poses of a ligand inside the active site of the protein. In this paper, we address the Molecular Docking search phase by formulating the problem in QUBO terms, suitable for an annealing approach. We propose a problem formulation as a weighted subgraph isomorphism between the ligand graph and the grid of the target protein pocket. In particular, we applied a graph representation to the ligand embedding all the geometrical properties of the molecule including its flexibility, and we created a weighted spatial grid to the 3D space region inside the pocket. Results and performance obtained with quantum annealers are compared with classical simulated annealing solvers.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript claims that the molecular docking search phase can be reformulated as a weighted subgraph isomorphism between a ligand graph (encoding 3D geometry and torsional flexibility) and a weighted spatial grid discretizing the protein pocket region; the resulting problem is cast as a QUBO and solved on quantum annealers, with performance compared against classical simulated annealing.
Significance. If the graph-plus-grid encoding can be shown to reproduce physically relevant binding poses and scores without post-hoc corrections, the work would constitute a concrete demonstration of quantum annealing applied to a high-dimensional combinatorial search problem in structural biology. The direct comparison to simulated annealing supplies a necessary classical baseline.
major comments (3)
- [Abstract / formulation] Abstract and formulation section: the central claim that the weighted subgraph isomorphism yields poses whose ranking matches physical binding affinity rests on the unshown assertion that a single set of edge weights on the ligand graph and pocket grid can encode all relevant energetic contributions. No derivation is supplied showing how hydrogen-bond, electrostatic, or desolvation terms are projected onto these weights; if they are omitted or approximated only geometrically, the QUBO optimum need not correspond to a low-RMSD or high-affinity pose.
- [Methods] Methods: the description of the ligand graph states that it 'embeds all the geometrical properties including its flexibility,' yet no explicit construction (vertex/edge attributes, distance or angle thresholds, or torsional variable encoding) is provided. Without this, it is impossible to verify that the resulting QUBO Hamiltonian is faithful to the original docking objective.
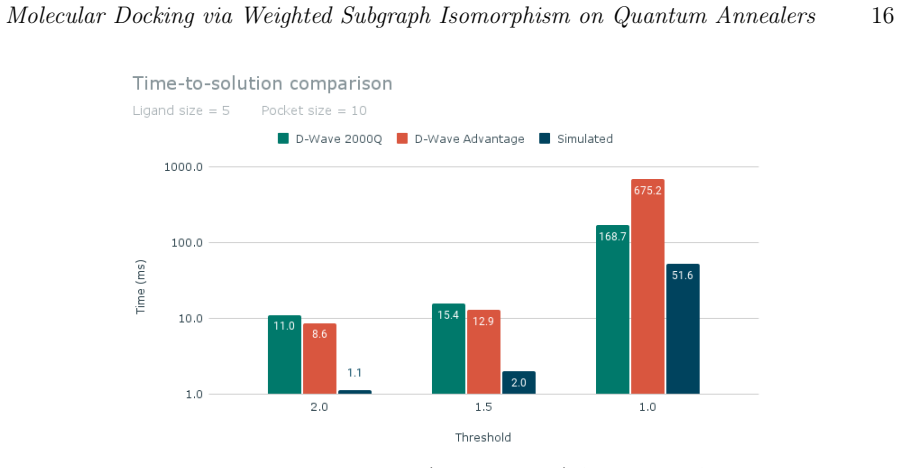
- [Results] Results: performance is compared to classical simulated annealing, but no quantitative metrics (RMSD distributions, enrichment factors, or success rates on a standard benchmark set such as PDBbind) are reported in the abstract or visible text. The absence of such validation data leaves the practical utility of the QUBO mapping untested.
minor comments (1)
- [Methods] Notation for the weighted grid and isomorphism objective should be introduced with explicit symbols rather than prose descriptions.
Simulated Author's Rebuttal
We thank the referee for the detailed and constructive report. We address each major comment below, with planned revisions to improve clarity on the scope and details of the formulation.
read point-by-point responses
-
Referee: [Abstract / formulation] Abstract and formulation section: the central claim that the weighted subgraph isomorphism yields poses whose ranking matches physical binding affinity rests on the unshown assertion that a single set of edge weights on the ligand graph and pocket grid can encode all relevant energetic contributions. No derivation is supplied showing how hydrogen-bond, electrostatic, or desolvation terms are projected onto these weights; if they are omitted or approximated only geometrically, the QUBO optimum need not correspond to a low-RMSD or high-affinity pose.
Authors: We agree that no derivation mapping full physical terms (hydrogen bonds, electrostatics, desolvation) onto the edge weights is provided. The formulation encodes geometric compatibility via distance-based weights between ligand atoms and grid points. We will revise the abstract and formulation section to explicitly qualify the approach as a geometric matching approximation and remove or qualify any implication that the QUBO optimum directly ranks by physical binding affinity. revision: yes
-
Referee: [Methods] Methods: the description of the ligand graph states that it 'embeds all the geometrical properties including its flexibility,' yet no explicit construction (vertex/edge attributes, distance or angle thresholds, or torsional variable encoding) is provided. Without this, it is impossible to verify that the resulting QUBO Hamiltonian is faithful to the original docking objective.
Authors: We acknowledge that the explicit construction details for the ligand graph (vertex/edge attributes, thresholds, and torsional encoding) are not supplied. In the revised Methods section we will add the precise definitions of vertices and edges, the distance and angle criteria used to define edges, and the binary variables introduced to represent torsional degrees of freedom. revision: yes
-
Referee: [Results] Results: performance is compared to classical simulated annealing, but no quantitative metrics (RMSD distributions, enrichment factors, or success rates on a standard benchmark set such as PDBbind) are reported in the abstract or visible text. The absence of such validation data leaves the practical utility of the QUBO mapping untested.
Authors: The reported results emphasize runtime and solution-quality comparisons on the QUBO objective itself rather than end-to-end docking benchmarks. We will expand the Results section to include RMSD values for the poses obtained on the example systems already presented and add an explicit limitations paragraph noting the lack of PDBbind-scale enrichment statistics. revision: partial
Circularity Check
No circularity: direct encoding presented without self-referential reductions
full rationale
The paper formulates molecular docking as a weighted subgraph isomorphism QUBO between a ligand graph and a protein pocket grid, then compares quantum and classical annealing solvers. No equations, fitted parameters, or predictions are shown that reduce by construction to inputs; the mapping is introduced as an explicit construction rather than a derived result. No self-citations, uniqueness theorems, or ansatzes appear in the provided text as load-bearing steps. The central claim is the encoding itself, which stands as an independent proposal without circular reduction to prior outputs or definitions within the paper.
Axiom & Free-Parameter Ledger
Forward citations
Cited by 1 Pith paper
-
A Physically-Informed Subgraph Isomorphism Approach to Molecular Docking Using Quantum Annealers
A novel QUBO formulation for quantum-annealer molecular docking adds physicochemical interaction terms to a prior geometric subgraph-isomorphism approach and reports improved accuracy on D-Wave devices.
Reference graph
Works this paper leans on
-
[1]
Morris and Marguerita Lim-Wilby.Molecular Docking, pages 365–382
Garrett M. Morris and Marguerita Lim-Wilby.Molecular Docking, pages 365–382. Humana Press, Totowa, NJ, 2008
work page 2008
-
[2]
Protein-liganddocking: Current status and future challenges
SergioFilipeSousa, PedroAlexandrinoFernandes, andMariaJoaoRamos. Protein-liganddocking: Current status and future challenges. Proteins:Structure, Function, and Bioinformatics , 65(1):15–26, 2006
work page 2006
-
[3]
Computationalmethodsforbiomoleculardocking
ThomasLengauerandMatthiasRarey. Computationalmethodsforbiomoleculardocking. Current Opinion in Structural Biology, 6(3):402–406, 1996
work page 1996
-
[4]
Paul C. D. Hawkins, A. Geoffrey Skillman, and Anthony Nicholls. Comparison of shape-matching and docking as virtual screening tools.Journal of Medicinal Chemistry, 50(1):74–82, 01 2007
work page 2007
-
[5]
Michael A. Nielsen and Isaac L. Chuang.Quantum Computation and Quantum Information: 10th Anniversary Edition. Cambridge University Press, 2010. Molecular Docking via Weighted Subgraph Isomorphism on Quantum Annealers 19
work page 2010
-
[6]
Henry A. Gabb, Richard M. Jackson, and Michael J.E. Sternberg. Modelling protein docking using shape complementarity, electrostatics and biochemical information11edited by j. thornton. Journal of Molecular Biology, 272(1):106–120, 1997
work page 1997
-
[7]
Caviar: a method for automatic cavity detection, description and decomposition into subcavities
Jean-Rémy Marchand, Bernard Pirard, Peter Ertl, and Finton Sirockin. Caviar: a method for automatic cavity detection, description and decomposition into subcavities. Journal of Computer-Aided Molecular Design, 35(6):737–750, 2021
work page 2021
-
[8]
G. Patrick Brady and Pieter F.W. Stouten. Fast prediction and visualization of protein binding pockets with pass. Journal of Computer-Aided Molecular Design, 14(4):383–401, May 2000
work page 2000
-
[9]
Jian Yu, Yong Zhou, Isao Tanaka, and Min Yao. Roll: a new algorithm for the detection of protein pockets and cavities with a rolling probe sphere.Bioinformatics, 26(1):46–52, 10 2009
work page 2009
-
[10]
Kevin Mato, Riccardo Mengoni, Daniele Ottaviani, and Gianluca Palermo. Quantum molecular unfolding. Quantum Science and Technology, 7(3):035020, jun 2022
work page 2022
-
[11]
Jinyin Zha, Jiaqi Su, Tiange Li, Chongyu Cao, Yin Ma, Hai Wei, Zhiguo Huang, Ling Qian, Kai Wen, and Jian Zhang. Encoding molecular docking for quantum computers.Journal of Chemical Theory and Computation, 19(24):9018–9024, 2023. PMID: 38090816
work page 2023
-
[12]
Molecular docking with gaussian boson sampling.Science Advances, 6(23):eaax1950, 2020
Leonardo Banchi, Mark Fingerhuth, Tomas Babej, Christopher Ing, and Juan Miguel Arrazola. Molecular docking with gaussian boson sampling.Science Advances, 6(23):eaax1950, 2020
work page 2020
-
[13]
Molecular docking via quantum approximate optimization algorithm, 2024
Qi-Ming Ding, Yi-Ming Huang, and Xiao Yuan. Molecular docking via quantum approximate optimization algorithm, 2024
work page 2024
-
[14]
A quantum algorithm for the sub-graph isomorphism problem, 2022
Nicola Mariella and Andrea Simonetto. A quantum algorithm for the sub-graph isomorphism problem, 2022
work page 2022
-
[15]
D.J.J.Marchand, M.Noori, A.Roberts, G.Rosenberg, B.Woods, U.Yildiz, M.Coons, D.Devore, and P. Margl. A variable neighbourhood descent heuristic for conformational search using a quantum annealer. Scientific Reports, 9(1):13708, Sep 2019
work page 2019
-
[16]
Barkoutsos, and Ivano Tavernelli
Stefano Mensa, Emre Sahin, Francesco Tacchino, Panagiotis Kl. Barkoutsos, and Ivano Tavernelli. Quantum machine learning framework for virtual screening in drug discovery: a prospective quantum advantage. CoRR, abs/2204.04017, 2022
-
[17]
Coarse-grained lattice protein folding on a quantum annealer, 2018
Tomas Babej, Christopher Ing, and Mark Fingerhuth. Coarse-grained lattice protein folding on a quantum annealer, 2018
work page 2018
-
[18]
Catherine C. McGeoch. Adiabatic Quantum Computation and Quantum Annealing: Theory and Practice. 2014
work page 2014
-
[19]
Siteferret: Beyond simple pocket identification in proteins
Luca Gagliardi and Walter Rocchia. Siteferret: Beyond simple pocket identification in proteins. Journal of Chemical Theory and Computation, 19(15):5242–5259, 2023. PMID: 37470784
work page 2023
-
[20]
Renxiao Wang, Xueliang Fang, Yipin Lu, Chao-Yie Yang, and Shaomeng Wang. The pdbbind database: Methodologies and updates.Journal of Medicinal Chemistry, 48(12):4111–4119, 2005
work page 2005
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.