Linear-Scaling Potential-Free Data-Driven Molecular Dynamics for Arbitrary-Sized Water Clusters (H₂O)_n
Pith reviewed 2026-05-23 08:14 UTC · model grok-4.3
The pith
A graph neural network predicts energies and forces for water clusters five times more accurately than DeepMD while scaling linearly.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
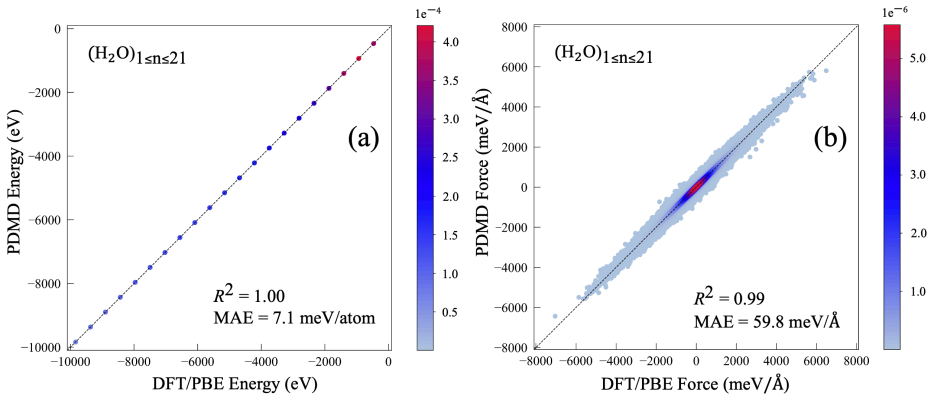
PDMD employs a Gaussian-based atomic geometry descriptor to generate high-dimensional equivariant features and ChemGNN to adaptively learn atomic chemical environments without a priori knowledge; after iterative self-consistent training the model achieves 1.39 meV/atom energy MAE and 50.7 meV/Å force MAE, outperforming DeepMD by factors of approximately five and three, respectively, and reproduces AIMD properties for clusters of thousands of molecules at orders-of-magnitude lower cost.
What carries the argument
ChemGNN, a graph neural network that takes Gaussian-based equivariant features and learns atomic chemical environments adaptively without requiring prior physical knowledge.
If this is right
- The linear-scaling property allows routine simulation of systems with thousands or more water molecules while retaining near-AIMD accuracy.
- Many-body effects that empirical force fields miss are now captured, enabling more reliable modeling of polyatomic systems.
- The supplied dataset of over 300,000 standardized water cluster structures can be used to benchmark other machine-learning MD methods.
- The same descriptor-plus-GNN architecture can be retrained on other molecular systems to produce general-purpose, potential-free MD engines.
Where Pith is reading between the lines
- Because the method needs no hand-crafted potentials, it could be retrained on mixtures or reactive systems where traditional force fields break down.
- The linear scaling opens the possibility of microsecond-scale trajectories for solvated biomolecules that remain inaccessible to AIMD.
- If the Gaussian descriptor proves transferable, the same workflow might replace empirical potentials in materials simulations beyond water.
Load-bearing premise
The combination of the Gaussian descriptor and ChemGNN captures every relevant many-body interaction for water clusters of any size, and the self-consistent training produces a model that generalizes outside the training structures.
What would settle it
Run PDMD and AIMD on a water cluster of 2000 molecules never seen in training and compare the resulting oxygen-oxygen radial distribution function or self-diffusion coefficient; a statistically significant mismatch would falsify the generalization claim.
Figures








read the original abstract
Conventional molecular dynamics (MD) simulation approaches, such as $\textit{ab initio}$ MD (AIMD) and empirical force field MD (EFFMD), face significant trade-offs between physical accuracy and computational efficiency. This work presents a linear-scaling potential-free data-driven molecular dynamics (PDMD) framework for predicting system energy and atomic forces of arbitrary-sized water clusters $(\text{H}_2\text{O})_n$. Specifically, PDMD employs a Gaussian-based atomic geometry descriptor to generate high-dimensional, equivariant features, then leverages ChemGNN, a graph neural network model that adaptively learns the atomic chemical environments without requiring $\textit{a priori}$ knowledge. Through an iterative self-consistent training approach, the converged PDMD achieves a mean absolute error of 1.39 meV/atom for energy and 50.7 meV/angstrom for forces, outperforming the state-of-the-art DeepMD by $\sim$5x in energy accuracy and $\sim$3x in force accuracy. As a result, the linear-scaling PDMD can reproduce the AIMD properties of water clusters at orders-of-magnitude lower computational cost, as illustrated by simulations of systems consisting of thousands or more molecules. These results demonstrate that the proposed PDMD offers multiphase predictive power and enables ultra-fast, general-purpose MD simulations while retaining AIMD-level accuracy. This accuracy is achieved by efficiently capturing many-body potentials that are critical in numerous polyatomic systems but are often missing in EFFMD. Moreover, we have constructed an $\textit{ab initio}$ dataset with over 300,000 $(\text{H}_2\text{O})_n$ structures, standardized in a unified PyTorch Geometric framework, to support scalable evaluation of artificial intelligence methods for molecular dynamics.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces a potential-free data-driven molecular dynamics (PDMD) framework for water clusters (H₂O)ₙ of arbitrary size. It employs a Gaussian-based atomic geometry descriptor to generate equivariant features and a graph neural network (ChemGNN) that learns atomic environments without a priori physical knowledge. Through iterative self-consistent training on an ab initio dataset of >300,000 structures, the converged model reports MAE of 1.39 meV/atom for energy and 50.7 meV/Å for forces, claiming ~5× and ~3× improvement over DeepMD, linear scaling, and the ability to reproduce AIMD properties for systems of thousands of molecules at orders-of-magnitude lower cost. A standardized PyTorch Geometric dataset is also released.
Significance. If the generalization claims hold, the work could enable AIMD-accurate MD simulations for large water systems at linear scaling and low cost, addressing key trade-offs in conventional methods while capturing many-body effects data-drivenly. The release of a large, standardized ab initio dataset in PyTorch Geometric is a clear strength that supports reproducibility and further method development in the field.
major comments (2)
- [Abstract] Abstract: The headline performance metrics (1.39 meV/atom energy, 50.7 meV/Å forces) and the central claim of applicability to arbitrary-sized clusters (including thousands of molecules) rest on unverified extrapolation. No information is supplied on the range of n in the >300k dataset, the train/test split, hyperparameter search, or any held-out validation on large-n systems independent of the training distribution.
- [Method description paragraph] Method description paragraph (iterative self-consistent training): The assumption that the Gaussian descriptor + ChemGNN, after iterative training, captures all relevant many-body interactions and generalizes to n far outside the training set is load-bearing for the linear-scaling claim but is not supported by any reported metrics on out-of-distribution cluster sizes or convergence diagnostics.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback. We address each major comment below and will revise the manuscript to provide the requested details and strengthen the presentation of generalization.
read point-by-point responses
-
Referee: [Abstract] Abstract: The headline performance metrics (1.39 meV/atom energy, 50.7 meV/Å forces) and the central claim of applicability to arbitrary-sized clusters (including thousands of molecules) rest on unverified extrapolation. No information is supplied on the range of n in the >300k dataset, the train/test split, hyperparameter search, or any held-out validation on large-n systems independent of the training distribution.
Authors: The abstract is necessarily concise, but the full manuscript describes the >300k structures as generated from ab initio calculations across a range of cluster sizes. We agree that explicit reporting is needed and will revise the abstract and add a new subsection in Methods detailing: (i) the distribution of n (spanning small clusters to several hundred molecules), (ii) the train/test split (80/20 with random sampling stratified by size), (iii) hyperparameter search procedure, and (iv) additional held-out tests on large-n clusters outside the primary training distribution. The applicability claim rests on the local, size-independent atomic descriptors and GNN architecture rather than global system size; we already demonstrate MD on systems of thousands of molecules, but will add explicit OOD metrics in the revision. revision: yes
-
Referee: [Method description paragraph] Method description paragraph (iterative self-consistent training): The assumption that the Gaussian descriptor + ChemGNN, after iterative training, captures all relevant many-body interactions and generalizes to n far outside the training set is load-bearing for the linear-scaling claim but is not supported by any reported metrics on out-of-distribution cluster sizes or convergence diagnostics.
Authors: The self-consistent iterative training alternates between model prediction and retraining on the growing dataset until energy/force errors stabilize, allowing the model to capture many-body effects present in the ab initio data without explicit physical priors. The Gaussian descriptor is strictly local (cutoff-based) and the ChemGNN operates on per-atom graphs, making the learned mapping size-independent by construction and thereby enabling linear scaling. We report final test-set MAEs after convergence but acknowledge the value of explicit OOD diagnostics; the revision will include training-convergence curves and, where data permits, performance on held-out larger clusters. revision: yes
Circularity Check
No significant circularity; performance from held-out dataset evaluation
full rationale
The paper constructs an ab initio dataset of >300k (H2O)n structures, trains ChemGNN on Gaussian descriptors via iterative self-consistent training, and reports MAE (1.39 meV/atom energy, 50.7 meV/Å forces) on evaluation data while comparing to DeepMD. These are standard empirical metrics from training/testing splits, not equations or claims that reduce to fitted inputs by construction. No self-citation load-bearing steps, uniqueness theorems, or ansatzes appear in the abstract or described method. The linear-scaling claim follows directly from the GNN architecture applied to arbitrary n, with no self-referential reduction.
Axiom & Free-Parameter Ledger
free parameters (2)
- ChemGNN architecture hyperparameters
- Gaussian descriptor width and cutoff parameters
axioms (2)
- domain assumption Atomic chemical environments can be learned adaptively from geometry descriptors without a priori physical rules
- ad hoc to paper Iterative self-consistent training converges to a stable, generalizable force field
invented entities (2)
-
PDMD framework
no independent evidence
-
ChemGNN
no independent evidence
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
PDMD employs a Gaussian-based atomic geometry descriptor ... ChemGNN ... iterative self-consistent training ... MAE 7.1 meV/atom ... (H2O)1≤n≤21
-
IndisputableMonolith/Foundation/RealityFromDistinction.leanreality_from_one_distinction unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
linear-scaling ... arbitrary-sized water clusters ... thousands or more molecules
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
author Alder, B. J. & author Wainwright, T. E. title Studies in molecular dynamics. i. general method . journal The Journal of Chemical Physics volume 31 , pages 459--466 ( year 1959 )
work page 1959
-
[2]
author Karplus, M. & author McCammon, J. A. title Molecular dynamics simulations of biomolecules . journal Nature structural biology volume 9 , pages 646--652 ( year 2002 )
work page 2002
-
[3]
author Mouvet, F. , author Villard, J. , author Bolnykh, V. & author Rothlisberger, U. title Recent advances in first-principles based molecular dynamics . journal Accounts of Chemical Research volume 55 , pages 221--230 ( year 2022 )
work page 2022
-
[4]
author Lifson, S. & author Warshel, A. title Consistent force field for calculations of conformations, vibrational spectra, and enthalpies of cycloalkane and n-alkane molecules . journal The Journal of Chemical Physics volume 49 , pages 5116--5129 ( year 1968 )
work page 1968
-
[5]
author Warshel, A. , author Kato, M. & author Pisliakov, A. V. title Polarizable force fields: history, test cases, and prospects . journal Journal of Chemical Theory and Computation volume 3 , pages 2034--2045 ( year 2007 )
work page 2034
-
[6]
author Iftimie, R. , author Minary, P. & author Tuckerman, M. E. title Ab initio molecular dynamics: Concepts, recent developments, and future trends . journal Proceedings of the National Academy of Sciences volume 102 , pages 6654--6659 ( year 2005 )
work page 2005
-
[7]
title An undulatory theory of the mechanics of atoms and molecules
author Schr \"o dinger, E. title An undulatory theory of the mechanics of atoms and molecules . journal Physical review volume 28 , pages 1049 ( year 1926 )
work page 1926
-
[8]
author Feynman, R. P. title Forces in molecules . journal Physical review volume 56 , pages 340 ( year 1939 )
work page 1939
-
[9]
author Hohenberg, P. & author Kohn, W. title Inhomogeneous electron gas . journal Physical review volume 136 , pages B864 ( year 1964 )
work page 1964
-
[10]
author Momany, F. , author McGuire, R. F. , author Burgess, A. & author Scheraga, H. A. title Energy parameters in polypeptides. vii. geometric parameters, partial atomic charges, nonbonded interactions, hydrogen bond interactions, and intrinsic torsional potentials for the naturally occurring amino acids . journal The Journal of Physical Chemistry volume...
work page 1975
-
[11]
author Best, R. B. et al. title Optimization of the additive charmm all-atom protein force field targeting improved sampling of the backbone , and side-chain 1 and 2 dihedral angles . journal Journal of chemical theory and computation volume 8 , pages 3257--3273 ( year 2012 )
work page 2012
-
[12]
author Tian, C. et al. title ff19sb: amino-acid-specific protein backbone parameters trained against quantum mechanics energy surfaces in solution . journal Journal of chemical theory and computation volume 16 , pages 528--552 ( year 2019 )
work page 2019
-
[13]
author Reif, M. M. , author Hünenberger, P. H. & author Oostenbrink, C. title New interaction parameters for charged amino acid side chains in the gromos force field . journal Journal of chemical theory and computation volume 8 , pages 3705--3723 ( year 2012 )
work page 2012
-
[14]
author Harder, E. et al. title Opls3: a force field providing broad coverage of drug-like small molecules and proteins . journal Journal of chemical theory and computation volume 12 , pages 281--296 ( year 2016 )
work page 2016
-
[15]
author Shi, Y. et al. title Polarizable atomic multipole-based amoeba force field for proteins . journal Journal of chemical theory and computation volume 9 , pages 4046--4063 ( year 2013 )
work page 2013
-
[16]
author Huang, L. & author Roux, B. title Automated force field parameterization for nonpolarizable and polarizable atomic models based on ab initio target data . journal Journal of chemical theory and computation volume 9 , pages 3543--3556 ( year 2013 )
work page 2013
-
[17]
author Fr \"o hlking, T. , author Bernetti, M. , author Calonaci, N. & author Bussi, G. title Toward empirical force fields that match experimental observables . journal The Journal of chemical physics volume 152 ( year 2020 )
work page 2020
-
[18]
author Senftle, T. P. et al. title The reaxff reactive force-field: development, applications and future directions . journal npj Computational Materials volume 2 , pages 1--14 ( year 2016 )
work page 2016
-
[19]
author Wu, Y. , author Chen, H. , author Wang, F. , author Paesani, F. & author Voth, G. A. title An improved multistate empirical valence bond model for aqueous proton solvation and transport . journal The Journal of Physical Chemistry B volume 112 , pages 467--482 ( year 2008 )
work page 2008
- [20]
-
[21]
author No \'e , F. , author Tkatchenko, A. , author M \"u ller, K.-R. & author Clementi, C. title Machine learning for molecular simulation . journal Annual review of physical chemistry volume 71 , pages 361--390 ( year 2020 )
work page 2020
-
[22]
author Mont \'a ns, F. J. , author Chinesta, F. , author G \'o mez-Bombarelli, R. & author Kutz, J. N. title Data-driven modeling and learning in science and engineering . journal Comptes Rendus M \'e canique volume 347 , pages 845--855 ( year 2019 )
work page 2019
-
[23]
title Electron correlation techniques in quantum chemistry: Recent advances
author Raghavachari, K. title Electron correlation techniques in quantum chemistry: Recent advances . journal Annual Review of Physical Chemistry volume 42 , pages 615--642 ( year 1991 )
work page 1991
-
[24]
author McMillan, W. L. title Ground state of liquid he 4 . journal Physical Review volume 138 , pages A442 ( year 1965 )
work page 1965
-
[25]
author Ross, I. G. title Calculations of the energy levels of acetylene by the method of antisymmetric molecular orbitals, including - interaction . journal Transactions of the Faraday Society volume 48 , pages 973--991 ( year 1952 )
work page 1952
-
[26]
author Pfau, D. , author Spencer, J. S. , author Matthews, A. G. & author Foulkes, W. M. C. title Ab initio solution of the many-electron schr \"o dinger equation with deep neural networks . journal Physical review research volume 2 , pages 033429 ( year 2020 )
work page 2020
-
[27]
author Kochkov, D. et al. title Machine learning--accelerated computational fluid dynamics . journal Proceedings of the National Academy of Sciences volume 118 , pages e2101784118 ( year 2021 )
work page 2021
-
[28]
author Glowinski, R. & author Pironneau, O. title Finite element methods for navier-stokes equations . journal Annual review of fluid mechanics volume 24 , pages 167--204 ( year 1992 )
work page 1992
-
[29]
author Li, Y. et al. title Ai-assisted superresolution cosmological simulations . journal Proceedings of the National Academy of Sciences volume 118 , pages e2022038118 ( year 2021 )
work page 2021
-
[30]
author Unke, O. T. et al. title Machine learning force fields . journal Chemical Reviews volume 121 , pages 10142--10186 ( year 2021 )
work page 2021
-
[31]
o lkopf, B. , author Smola, A. & author M \
author Sch \"o lkopf, B. , author Smola, A. & author M \"u ller, K.-R. title Nonlinear component analysis as a kernel eigenvalue problem . journal Neural computation volume 10 , pages 1299--1319 ( year 1998 )
work page 1998
-
[32]
author Fukushima, K. title Neocognitron: A self-organizing neural network model for a mechanism of pattern recognition unaffected by shift in position . journal Biological cybernetics volume 36 , pages 193--202 ( year 1980 )
work page 1980
-
[33]
title Visual feature extraction by a multilayered network of analog threshold elements
author Fukushima, K. title Visual feature extraction by a multilayered network of analog threshold elements . journal IEEE Transactions on Systems Science and Cybernetics volume 5 , pages 322--333 ( year 1969 )
work page 1969
-
[34]
author Morawietz, T. , author Singraber, A. , author Dellago, C. & author Behler, J. title How van der waals interactions determine the unique properties of water . journal Proceedings of the National Academy of Sciences volume 113 , pages 8368--8373 ( year 2016 )
work page 2016
-
[35]
author Becke, A. D. title Density-functional exchange-energy approximation with correct asymptotic behavior . journal Physical review A volume 38 , pages 3098 ( year 1988 )
work page 1988
-
[36]
title Atom-centered symmetry functions for constructing high-dimensional neural network potentials
author Behler, J. title Atom-centered symmetry functions for constructing high-dimensional neural network potentials . journal The Journal of chemical physics volume 134 ( year 2011 )
work page 2011
-
[37]
author Smith, J. S. , author Isayev, O. & author Roitberg, A. E. title Ani-1: an extensible neural network potential with dft accuracy at force field computational cost . journal Chemical science volume 8 , pages 3192--3203 ( year 2017 )
work page 2017
-
[38]
author Sch \"u tt, K. et al. title Schnet: A continuous-filter convolutional neural network for modeling quantum interactions . journal Advances in neural information processing systems volume 30 ( year 2017 )
work page 2017
-
[39]
author Scarselli, F. , author Gori, M. , author Tsoi, A. C. , author Hagenbuchner, M. & author Monfardini, G. title The graph neural network model . journal IEEE transactions on neural networks volume 20 , pages 61--80 ( year 2008 )
work page 2008
-
[40]
author Hasebe, T. title Knowledge-embedded message-passing neural networks: improving molecular property prediction with human knowledge . journal ACS omega volume 6 , pages 27955--27967 ( year 2021 )
work page 2021
-
[41]
author Park, C. W. et al. title Accurate and scalable graph neural network force field and molecular dynamics with direct force architecture . journal npj Computational Materials volume 7 , pages 73 ( year 2021 )
work page 2021
-
[42]
author Choudhary, K. et al. title Unified graph neural network force-field for the periodic table: solid state applications . journal Digital Discovery volume 2 , pages 346--355 ( year 2023 )
work page 2023
- [43]
-
[44]
author Keutsch, F. N. & author Saykally, R. J. title Water clusters: Untangling the mysteries of the liquid, one molecule at a time . journal Proceedings of the National Academy of Sciences volume 98 , pages 10533--10540 ( year 2001 )
work page 2001
-
[45]
author Rognoni, A. , author Conte, R. & author Ceotto, M. title How many water molecules are needed to solvate one? journal Chemical Science volume 12 , pages 2060--2064 ( year 2021 )
work page 2060
-
[46]
author Brubach, J.-B. , author Mermet, A. , author Filabozzi, A. , author Gerschel, A. & author Roy, P. title Signatures of the hydrogen bonding in the infrared bands of water . journal The Journal of chemical physics volume 122 ( year 2005 )
work page 2005
-
[47]
author Lee, C. , author Yang, W. & author Parr, R. G. title Development of the colle-salvetti correlation-energy formula into a functional of the electron density . journal Physical review B volume 37 , pages 785 ( year 1988 )
work page 1988
-
[48]
author Perdew, J. P. , author Burke, K. & author Ernzerhof, M. title Generalized gradient approximation made simple . journal Physical review letters volume 77 , pages 3865 ( year 1996 )
work page 1996
-
[49]
author Zeng, J. et al. title Deepmd-kit v2: A software package for deep potential models . journal The Journal of Chemical Physics volume 159 ( year 2023 )
work page 2023
-
[50]
author Wang, Y. , author Babin, V. , author Bowman, J. M. & author Paesani, F. title The water hexamer: cage, prism, or both. full dimensional quantum simulations say both . journal Journal of the American Chemical Society volume 134 , pages 11116--11119 ( year 2012 )
work page 2012
-
[51]
author Latimer, W. M. & author Rodebush, W. H. title Polarity and ionization from the standpoint of the lewis theory of valence. journal Journal of the American Chemical Society volume 42 , pages 1419--1433 ( year 1920 )
work page 1920
-
[52]
author Malloum, A. , author Fifen, J. J. , author Dhaouadi, Z. , author Engo, S. G. N. & author Conradie, J. title Structures, relative stability and binding energies of neutral water clusters,(h 2 o) 2--30 . journal New Journal of Chemistry volume 43 , pages 13020--13037 ( year 2019 )
work page 2019
-
[53]
author Silverstein, K. A. , author Haymet, A. & author Dill, K. A. title The strength of hydrogen bonds in liquid water and around nonpolar solutes . journal Journal of the American Chemical Society volume 122 , pages 8037--8041 ( year 2000 )
work page 2000
-
[54]
author Musaelian, A. et al. title Learning local equivariant representations for large-scale atomistic dynamics . journal Nature Communications volume 14 , pages 579 ( year 2023 )
work page 2023
-
[55]
author Wang, W. , author Wu, Z. , author Dietschreit, J. C. & author G \'o mez-Bombarelli, R. title Learning pair potentials using differentiable simulations . journal The Journal of Chemical Physics volume 158 ( year 2023 )
work page 2023
-
[56]
author Herman, K. M. , author Stone, A. J. & author Xantheas, S. S. title A classical model for three-body interactions in aqueous ionic systems . journal The Journal of Chemical Physics volume 157 ( year 2022 )
work page 2022
-
[57]
author Xie, S. R. , author Rupp, M. & author Hennig, R. G. title Ultra-fast interpretable machine-learning potentials . journal npj Computational Materials volume 9 , pages 162 ( year 2023 )
work page 2023
-
[58]
author Swanson, J. M. et al. title Proton solvation and transport in aqueous and biomolecular systems: Insights from computer simulations . journal The Journal of Physical Chemistry B volume 111 , pages 4300--4314 ( year 2007 )
work page 2007
-
[59]
author Simons, J. title Molecular anions . journal The Journal of Physical Chemistry A volume 112 , pages 6401--6511 ( year 2008 )
work page 2008
-
[60]
title Systematic analysis of many-body interactions in molecular solids
author Jansen, L. title Systematic analysis of many-body interactions in molecular solids . journal Physical Review volume 125 , pages 1798 ( year 1962 )
work page 1962
-
[61]
author Ejtehadi, M. , author Avall, S. & author Plotkin, S. title Three-body interactions improve the prediction of rate and mechanism in protein folding models . journal Proceedings of the National Academy of Sciences volume 101 , pages 15088--15093 ( year 2004 )
work page 2004
-
[62]
author Konstantinova, E. V. & author Skorobogatov, V. A. title Application of hypergraph theory in chemistry . journal Discrete Mathematics volume 235 , pages 365--383 ( year 2001 )
work page 2001
-
[63]
author Jost, J. & author Mulas, R. title Hypergraph laplace operators for chemical reaction networks . journal Advances in mathematics volume 351 , pages 870--896 ( year 2019 )
work page 2019
-
[64]
author K \"u hne, T. D. et al. title Cp2k: An electronic structure and molecular dynamics software package-quickstep: Efficient and accurate electronic structure calculations . journal The Journal of Chemical Physics volume 152 ( year 2020 )
work page 2020
-
[65]
author Goedecker, S. , author Teter, M. & author Hutter, J. title Separable dual-space gaussian pseudopotentials . journal Physical Review B volume 54 , pages 1703 ( year 1996 )
work page 1996
-
[66]
author Woon, D. E. & author Dunning Jr, T. H. title Gaussian basis sets for use in correlated molecular calculations. iv. calculation of static electrical response properties . journal The Journal of chemical physics volume 100 , pages 2975--2988 ( year 1994 )
work page 1994
-
[67]
author Genovese, L. , author Deutsch, T. , author Neelov, A. , author Goedecker, S. & author Beylkin, G. title Efficient solution of poisson’s equation with free boundary conditions . journal The Journal of chemical physics volume 125 ( year 2006 )
work page 2006
-
[68]
author Zhao, Y. & author Truhlar, D. G. title The m06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four m06-class functionals and 12 other functionals . journal Theoretical chemistry accounts volume 120 ...
work page 2008
-
[69]
author De, S. , author Bart \'o k, A. P. , author Cs \'a nyi, G. & author Ceriotti, M. title Comparing molecules and solids across structural and alchemical space . journal Physical Chemistry Chemical Physics volume 18 , pages 13754--13769 ( year 2016 )
work page 2016
-
[70]
author Laakso, J. et al. title Updates to the dscribe library: New descriptors and derivatives . journal The Journal of Chemical Physics volume 158 ( year 2023 )
work page 2023
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.