A ReaxFF-based thermomechanical analysis of N-carbophenes: phase-change, thermal expansion, and high temperature synthesis pathway
Pith reviewed 2026-05-15 20:58 UTC · model grok-4.3
The pith
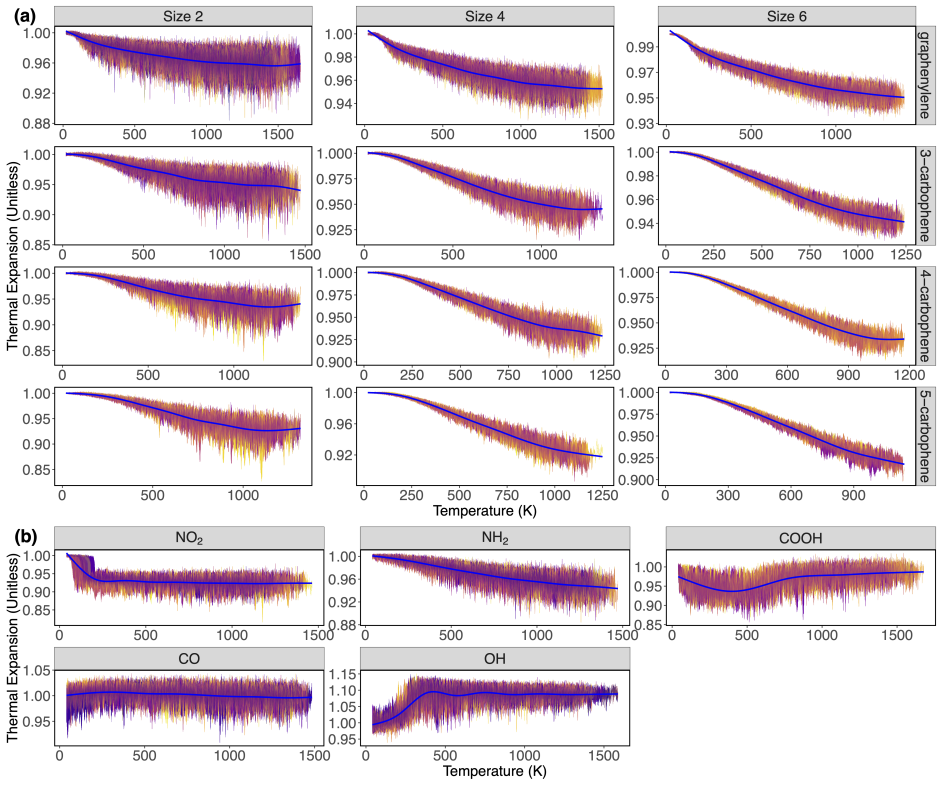
N-carbophenes stay stable above 1000 K, with phase-change temperatures falling as phenylene chain length grows due to antiaromaticity.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Using ReaxFF-based reactive molecular dynamics simulations with temperature-ramp protocols and statistical checks, the authors demonstrate that N-carbophenes remain stable above 1000 K. Phase-change onset temperatures decrease with increasing N-phenylene chain length in the pristine materials because of rising antiaromaticity in the central phenylene segments. Pristine N-carbophenes show negative area thermal expansion while functional groups shift the behavior toward positive or keep it negative; the groups themselves stay bonded well past the transition. A temperature-induced conversion from graphenylene (2-carbophene) to gamma-graphyne is shown to be accessible.
What carries the argument
ReaxFF reactive molecular dynamics simulations that track bonding energetics, structural rearrangements, and area changes during controlled temperature ramps on both pristine and functionalized N-carbophenes.
If this is right
- Upper temperature limits above 1000 K can be used to set safe operating ranges for N-carbophene-based gas-storage or electronic devices.
- Phase-change onset can be shifted by selecting different N-phenylene chain lengths in the pristine structures.
- Thermal expansion sign can be switched from negative to positive by adding suitable functional groups.
- Functional groups remain attached after the material has passed its phase-change temperature.
- Graphenylene can be converted to gamma-graphyne by a purely thermal route.
Where Pith is reading between the lines
- The antiaromaticity explanation for earlier phase changes may apply to other 2D carbon lattices that contain similar ring sequences, suggesting a broader design rule for tuning thermal stability.
- Laboratory attempts to heat graphenylene films or powders through the simulated transition range could test whether the gamma-graphyne product forms in practice.
- The ability to flip the sign of thermal expansion with functional groups opens the possibility of engineering N-carbophene composites that show near-zero net expansion over wide temperature intervals.
- High-temperature stability combined with a built-in conversion pathway positions these frameworks as possible precursors for making other graphyne-like sheets under controlled heating conditions.
Load-bearing premise
The ReaxFF force field parameters reproduce the bonding energies, phase transitions, and thermal expansion of N-carbophenes across the full temperature window examined.
What would settle it
An experiment that records decomposition or a phase change in any N-carbophene below roughly 900 K, or that fails to produce gamma-graphyne when graphenylene is heated through the predicted window, would falsify the stability and transition claims.
Figures





read the original abstract
N-carbophenes are a class of two-dimensional covalent organic frameworks with potential for solid-state gas storage and as 2D topological materials. Previous studies have demonstrated that variations in their bonding, topology, and functionalization enable the tuning of their chemical, electrical, and mechanical properties. Yet, the thermal stability and high-temperature behavior of pristine and functionalized N-carbophenes remain unexplored. Using ReaxFF-based reactive molecular dynamics (RMD) simulations with extensive statistical validation, we performed temperature-ramp MD simulations of pristine and functionalized N-carbophenes. We demonstrate that N-carbophenes remain stable up to temperatures above 1000 K. The phase-change onset temperatures decrease as the N-phenylene chain length increases in pristine N-carbophenes, attributed to increasing antiaromaticity in the central phenylene segments, thereby contributing to the foundational understanding of aromatic versus antiaromatic bonding in 2D carbon networks, a topic of considerable interest in theoretical chemistry. Pristine N-carbophenes exhibit negative area thermal expansion (NATE), whereas functional groups modulate this, leading to either negative or positive expansion. Functional groups remain stably bonded well above the transition temperature. We also show that a temperature-induced phase transition from graphenylene (2-carbophene) to {\gamma}-graphyne is possible. Our results provide upper bounds on N-carbophene stability, clarify the relationships between structure and thermal properties, and identify a new transformation pathway. These results will have applications in tunable band gaps, porous architectures, or chemically accessible sites.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript reports ReaxFF-based reactive molecular dynamics simulations of pristine and functionalized N-carbophenes, claiming thermal stability above 1000 K, decreasing phase-change onset temperatures with increasing N-phenylene chain length due to antiaromaticity, negative area thermal expansion modulated by functional groups, stable bonding of functional groups above transition temperatures, and a temperature-induced transition from graphenylene (2-carbophene) to γ-graphyne.
Significance. If the ReaxFF force field is shown to be transferable to these N-doped 2D frameworks, the work supplies useful upper bounds on thermal stability and structure-property relations for 2D COFs, including a potential high-temperature synthesis route to γ-graphyne. The reactive MD approach for exploring high-T transformations is a methodological strength, but the lack of any reported numerical values, error bars, or cross-validation against DFT or experiment for onset temperatures and expansion coefficients limits the immediate significance.
major comments (3)
- [Abstract] Abstract: the assertion of 'extensive statistical validation' is unsupported by any numerical data, error bars, system sizes, or validation metrics against experiment or higher-level theory, which is load-bearing for the central stability and trend claims.
- [Results] Results (phase-change section): the attribution of decreasing onset temperatures to increasing antiaromaticity in central phenylene segments is presented without quantitative support such as NICS values, bond-length alternation metrics, or comparative energy decompositions from the trajectories.
- [Methods] Methods: no information is given on the specific ReaxFF parameterization for N-carbophenes, training data relevance to antiaromatic segments or high-T reconstructions, or any benchmark comparisons of predicted onset temperatures and thermal expansion coefficients against DFT or experiment.
minor comments (1)
- [Abstract] Abstract: the acronym NATE is used without definition on first appearance.
Simulated Author's Rebuttal
We thank the referee for the constructive and detailed feedback on our manuscript. We have carefully reviewed each major comment and provide point-by-point responses below, indicating where revisions will be made to strengthen the presentation of our ReaxFF results.
read point-by-point responses
-
Referee: [Abstract] Abstract: the assertion of 'extensive statistical validation' is unsupported by any numerical data, error bars, system sizes, or validation metrics against experiment or higher-level theory, which is load-bearing for the central stability and trend claims.
Authors: We agree that the abstract claim requires explicit supporting details. In the revised manuscript we will add concrete information on system sizes (supercells containing 800–2000 atoms), the number of independent temperature-ramp runs (minimum of five per structure for statistical averaging), standard-deviation error bars on onset temperatures and area-expansion coefficients, and internal consistency metrics such as total-energy drift and root-mean-square atomic displacements extracted directly from the trajectories. These additions will be cross-referenced in the Results and Methods sections. revision: yes
-
Referee: [Results] Results (phase-change section): the attribution of decreasing onset temperatures to increasing antiaromaticity in central phenylene segments is presented without quantitative support such as NICS values, bond-length alternation metrics, or comparative energy decompositions from the trajectories.
Authors: We acknowledge that the current link to antiaromaticity remains qualitative. The revised Results section will include quantitative bond-length alternation (BLA) values averaged over the central phenylene rings from equilibrated trajectory segments at multiple temperatures. In addition, representative fragments will be extracted and subjected to DFT single-point calculations to obtain NICS(0) and NICS(1) values, together with a ReaxFF energy decomposition isolating the contribution of the central segments. These data will be presented in a new supplementary figure and table. revision: yes
-
Referee: [Methods] Methods: no information is given on the specific ReaxFF parameterization for N-carbophenes, training data relevance to antiaromatic segments or high-T reconstructions, or any benchmark comparisons of predicted onset temperatures and thermal expansion coefficients against DFT or experiment.
Authors: We will expand the Methods section to specify the exact ReaxFF parameter set (C/N/H parameters from the 2019 parameterization of Kowalik et al.), its training-set coverage of aromatic/antiaromatic rings and bond-dissociation events, and the simulation protocols (NVT ensemble, 0.25 fs timestep, Nosé-Hoover thermostat). We will also add a short paragraph clarifying that full-scale DFT benchmarks for high-temperature onset temperatures are outside the scope of the present reactive-MD study owing to system size; however, we will include limited DFT geometry optimizations on small model clusters to confirm local bonding motifs and will reference prior ReaxFF validation studies on related 2D carbon frameworks. revision: partial
Circularity Check
No significant circularity; results are direct outputs of ReaxFF MD trajectories
full rationale
The paper reports stability limits, phase-change onset temperatures, NATE behavior, and a graphenylene-to-γ-graphyne transition as direct results of temperature-ramp reactive molecular dynamics simulations. No equations, fitted parameters, or self-citations within the study reduce these observables to quantities defined or fitted by the same work. The ReaxFF parameterization is cited from prior literature (including co-author van Duin), but this constitutes standard method application rather than a load-bearing self-citation chain that forces the reported trends. The attribution of onset-temperature dependence to antiaromaticity is an interpretive claim, not a mathematical equivalence. The derivation chain remains self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption ReaxFF reactive force field accurately captures bond breaking, formation, and thermomechanical response in N-carbophenes up to and beyond 1000 K.
Reference graph
Works this paper leans on
-
[1]
A. P. Côté, A. I. Benin, N. W. Ockwig, M. O’Keeffe, A. J. Matzger, O. M. Yaghi, Porous, crystalline, covalent organic frameworks, Science 310 (5751) (2005) 1166–1170.arXiv:http://science.sciencemag.org/ content/310/5751/1166.full.pdf,doi:10.1126/science.1120411. URLhttp://science.sciencemag.org/content/310/5751/1166
-
[2]
C. Wang, Z. Zhang, Y . Zhu, C. Yang, J. Wu, W. Hu, 2d covalent organic frameworks: From synthetic strategies to advanced optical-electrical-magnetic functionalities, Advanced Materials 34 (17) (2022) 2102290. arXiv:https://advanced-onlinelibrary-wiley-com.eres.library.manoa.hawaii.edu/doi/ pdf/10.1002/adma.202102290,doi:https://doi-org.eres.library.manoa....
-
[3]
T. Zhang, G. Zhang, L. Chen, 2d conjugated covalent organic frameworks: Defined synthesis and tailor-made functions, Accounts of Chemical Research 55 (6) (2022) 795–808, pMID: 35025209.arXiv:https://doi. org/10.1021/acs.accounts.1c00693,doi:10.1021/acs.accounts.1c00693. URLhttps://doi.org/10.1021/acs.accounts.1c00693
-
[4]
X. Ni, H. Li, F. Liu, J.-L. Brédas, Engineering of flat bands and dirac bands in two-dimensional covalent organic frameworks (cofs): relationships among molecular orbital symmetry, lattice symmetry, and electronic-structure characteristics, Mater. Horiz. 9 (2022) 88–98.doi:10.1039/D1MH00935D. URLhttp://dx.doi.org/10.1039/D1MH00935D
-
[5]
S. Fu, J. Zhang, X. Li, E. Jin, L. Gao, R. Dong, Z. Wang, X. Feng, H. I. Wang, M. Bonn, Fundamentals of charge transport in two-dimensional framework materials, Nature Reviews Materials (2025) 1–22doi:10. 1038/s41578-025-00840-z. URLhttps://doi.org/10.1038/s41578-025-00840-z
-
[6]
S.-Y . Ding, M. Dong, Y .-W. Wang, Y .-T. Chen, H.-Z. Wang, C.-Y . Su, W. Wang, Thioether-based fluorescent covalent organic framework for selective detection and facile removal of mercury(ii), Journal of the American Chemical Society 138 (9) (2016) 3031–3037, pMID: 26878337.arXiv:https://doi.org/10.1021/jacs. 5b10754,doi:10.1021/jacs.5b10754. URLhttps://...
-
[7]
S. Mitra, H. S. Sasmal, T. Kundu, S. Kandambeth, K. Illath, D. Díaz Díaz, R. Banerjee, Targeted drug delivery in covalent organic nanosheets (cons) via sequential postsynthetic modification, Journal of the American Chemical Society 139 (12) (2017) 4513–4520, pMID: 28256830.arXiv:https://doi.org/10.1021/jacs.7b00925, doi:10.1021/jacs.7b00925. URLhttps://do...
-
[8]
S. Wang, Q. Wang, P. Shao, Y . Han, X. Gao, L. Ma, S. Yuan, X. Ma, J. Zhou, X. Feng, B. Wang, Exfoliation of covalent organic frameworks into few-layer redox-active nanosheets as cathode materials for lithium-ion batteries, Journal of the American Chemical Society 139 (12) (2017) 4258–4261, pMID: 28316238.arXiv: https://doi.org/10.1021/jacs.7b02648,doi:10...
-
[9]
J. Dong, Y . Wang, G. Liu, Y . Cheng, D. Zhao, Isoreticular covalent organic frameworks for hydrocarbon uptake and separation: the important role of monomer planarity, CrystEngComm 19 (2017) 4899–4904.doi:10. 1039/C7CE00344G. URLhttp://dx.doi.org/10.1039/C7CE00344G
-
[10]
M. Raju, P. B. Govindaraju, A. C. T. van Duin, M. Ihme, Atomistic and continuum scale modeling of func- tionalized graphyne membranes for water desalination, Nanoscale 10 (2018) 3969–3980.doi:10.1039/ C7NR07963J. URLhttp://dx.doi.org/10.1039/C7NR07963J
-
[11]
Q.-S. Du, P.-D. Tang, H.-L. Huang, F.-L. Du, K. Huang, N.-Z. Xie, S.-Y . Long, Y .-M. Li, J.-S. Qiu, R.-B. Huang, A new type of two-dimensional carbon crystal prepared from 1,3,5-trihydroxybenzene, Scientific Reports 7 (2017) 40796.doi:10.1038/srep40796. URLhttps://doi.org/10.1038/srep40796
-
[12]
C. E. Junkermeier, J. P. Luben, R. Paupitz, N-carbophenes: two-dimensional covalent organic frameworks de- rived from linear n-phenylenes, Materials Research Express 6 (11) (2019) 115103.doi:10.1088/2053-1591/ ab4513. URLhttps://doi.org/10.1088/2F2053-1591/2Fab4513
-
[13]
C. E. Junkermeier, G. Psofogiannakis, R. Paupitz, Covalent adsorption of functional groups on n-carbophenes, Materials Research Express (Feb 2022).doi:10.1088/2053-1591/ac4c19. URLhttps://doi.org/10.1088/2053-1591/ac4c19
-
[14]
N.-J. Yang, H. Yang, Z. Huang, J.-M. Zhang, Second-order topological insulators in kekulé-patterned hexag- onal biphenylene networks, Applied Physics Letters 126 (3) (2025) 033101.arXiv:https://pubs.aip. org/aip/apl/article-pdf/doi/10.1063/5.0239997/20355876/033101_1_5.0239997.pdf,doi:10. 1063/5.0239997. URLhttps://doi.org/10.1063/5.0239997
work page doi:10.1063/5.0239997/20355876/033101_1_5.0239997.pdf 2025
-
[15]
G. H. Batista, G. Psofogiannakis, C. E. Junkermeier, R. Paupitz, Mechanical properties and deformation-driven band gap tuning on [n]-carbophenes, Computational Materials Science 222 (2023) 112103.doi:https:// doi.org/10.1016/j.commatsci.2023.112103. URLhttps://www.sciencedirect.com/science/article/pii/S0927025623000976
-
[16]
M. Arockiaraj, D. Paul, M. Rahul, J. Clement, S. Tigga, K. Balasubramanian, Topological and entropy indices in qspr studies of n-carbophene covalent organic frameworks, BioNanoScience 14 (3) (2024) 2762–2773.doi: 10.1007/s12668-024-01546-2. URLhttps://doi.org/10.1007/s12668-024-01546-2
-
[17]
C. E. Junkermeier, E. Larmand, J.-C. Morais, J. Kobebel, K. Lavarez, R. M. Adra, J. Yang, V . A. Diaz, R. Paupitz, G. Psofogiannakis, Functionalized carbophenes as high-capacity versatile gas adsorbents: An ab initio study, Computational Materials Science 232 (2024) 112665.doi:https://doi.org/10.1016/j. 11 commatsci.2023.112665. URLhttps://www.sciencedire...
work page doi:10.1016/j 2024
-
[18]
C. E. Junkermeier, J. Kobebel, K. Lavarez, R. M. Adra, J. Yang, V . A. Diaz, R. Paupitz, G. Psofogiannaki, Density functional tight-binding derived data of gas capture in functionalized carbophenes, Data in Brief 55 (2024) 110652.doi:https://doi.org/10.1016/j.dib.2024.110652. URLhttps://www.sciencedirect.com/science/article/pii/S235234092400619X
-
[19]
M. Servalli, H. C. Öttinger, D. Schlüter, Synthesizing molecular fishing nets, Physics Today 71 (5) (2018) 40–47. doi:10.1063/PT.3.3921. URLhttps://physicstoday.scitation.org/doi/pdf/10.1063/PT.3.3921
-
[20]
T. P. Senftle, S. Hong, M. M. Islam, S. B. Kylasa, Y . Zheng, Y . K. Shin, C. Junkermeier, R. Engel-Herbert, M. J. Janik, H. M. Aktulga, T. Verstraelen, G. Ananth, A. C. van Duin, The reaxffreactive force-field: de- velopment, applications and future directions, npj Computational Materials 2 (2016) 15011.doi:10.1038/ npjcompumats.2015.11
work page 2016
-
[21]
SCM, Theoretical Chemistry, ReaxFF (2019). URLhttps://www.scm.com
work page 2019
-
[22]
G. J. Martyna, M. E. Tuckerman, D. J. Tobias, M. L. Klein, Explicit reversible integrators for extended systems dynamics, Molecular Physics 87 (5) (1996) 1117–1157
work page 1996
-
[23]
W. J. Mortier, S. K. Ghosh, S. Shankar, Electronegativity-equalization method for the calculation of atomic charges in molecules, Journal of the American Chemical Society 108 (15) (1986) 4315–4320.arXiv:https: //doi.org/10.1021/ja00275a013,doi:10.1021/ja00275a013. URLhttps://doi.org/10.1021/ja00275a013
-
[24]
M. Kowalik, C. M. Ashraf, A. van Duin, Atomistic investigation of a carbonization process for c/h/o/n-based polymers with use of the reactive potentials: Reaxff, in: APS March Meeting Abstracts, V ol. 2019, 2019, pp. X19–010
work page 2019
-
[25]
Z. Gao, J. Zhu, S. Rajabpour, K. Joshi, M. Kowalik, B. Croom, Y . Schwab, L. Zhang, C. Bumgardner, K. R. Brown, D. Burden, J. W. Klett, A. C. T. van Duin, L. V . Zhigilei, X. Li, Graphene reinforced carbon fibers, Sci- ence Advances 6 (17) (2020) eaaz4191.arXiv:https://www.science.org/doi/pdf/10.1126/sciadv. aaz4191,doi:10.1126/sciadv.aaz4191. URLhttps://...
-
[26]
Q. Mao, S. Rajabpour, M. Kowalik, A. C. van Duin, Predicting cost-effective carbon fiber precursors: Unravel- ing the functionalities of oxygen and nitrogen-containing groups during carbonization from reaxffsimulations, Carbon 159 (2020) 25–36.doi:https://doi.org/10.1016/j.carbon.2019.12.008. URLhttps://www.sciencedirect.com/science/article/pii/S0008622319312436
-
[27]
Schrödinger, LLC, The PyMOL molecular graphics system, version 1.8, a (November 2015)
work page 2015
-
[28]
W. Humphrey, A. Dalke, K. Schulten, Vmd: Visual molecular dynamics, Journal of Molecular Graphics 14 (1) (1996) 33–38.doi:https://doi.org/10.1016/0263-7855(96)00018-5. URLhttps://www.sciencedirect.com/science/article/pii/0263785596000185
-
[29]
Y . Ishigaki, T. Shimajiri, T. Takeda, R. Katoono, T. Suzuki, Longest c–c single bond among neutral hydrocarbons with a bond length beyond 1.8 Å, Chem 4 (4) (2018) 795–806.doi:https://doi.org/10.1016/j.chempr. 2018.01.011. URLhttps://www.sciencedirect.com/science/article/pii/S2451929418300330
-
[30]
A. Kassambara, rstatix: Pipe-Friendly Framework for Basic Statistical Tests, r package version 0.7.2 (2023). URLhttps://CRAN.R-project.org/package=rstatix 12
work page 2023
-
[31]
A. Trapletti, K. Hornik, tseries: Time Series Analysis and Computational Finance, r package version 0.10-52. (2022). URLhttps://CRAN.R-project.org/package=tseries
work page 2022
-
[32]
J. V . Bradley, Distribution-free statistical tests, Tech. Rep. W ADD TECHNICAL REPORT 60-661, United States Air Force (1960)
work page 1960
-
[33]
W. M. Patefield, Algorithm as 159: An efficient method of generating random r×c tables with given row and column totals, Journal of the Royal Statistical Society. Series C (Applied Statistics) 30 (1) (1981) 91–97. doi:10.2307/2346669. URLhttp://www.jstor.org/stable/2346669
-
[34]
A. Schleifenbaum, N. Feeder, K. C. V ollhardt, The x-ray crystal structure of linear [3]phenylene, Tetrahedron Letters 42 (42) (2001) 7329 – 7332.doi:https://doi.org/10.1016/S0040-4039(01)01425-3. URLhttp://www.sciencedirect.com/science/article/pii/S0040403901014253
-
[35]
S. P. Couch, A. P. Bray, C. Ismay, E. Chasnovski, B. S. Baumer, M. Çetinkaya Rundel, infer: An R package for tidyverse-friendly statistical inference, Journal of Open Source Software 6 (65) (2021) 3661.doi:10.21105/ joss.03661
work page 2021
-
[36]
Wilkins, treemapify: Draw Treemaps in ’ggplot2’, r package version 2.5.5 (2021)
D. Wilkins, treemapify: Draw Treemaps in ’ggplot2’, r package version 2.5.5 (2021). URLhttps://CRAN.R-project.org/package=treemapify
work page 2021
-
[37]
T. L. Pedersen, patchwork: The Composer of Plots, r package version 1.2.0 (2024). URLhttps://CRAN.R-project.org/package=patchwork
work page 2024
-
[38]
T. Barron, J. Barton, J. Johnson, On the stability of some polycyclic biphenylene derivatives, Tetrahedron 22 (8) (1966) 2609 – 2613.doi:https://doi.org/10.1016/S0040-4020(01)99053-2. URLhttp://www.sciencedirect.com/science/article/pii/S0040402001990532
-
[39]
Gutman, Easy method for the calculation of the algebraic structure count of phenylenes, J
I. Gutman, Easy method for the calculation of the algebraic structure count of phenylenes, J. Chem. Soc., Faraday Trans. 89 (1993) 2413–2416.doi:10.1039/FT9938902413. URLhttp://dx.doi.org/10.1039/FT9938902413
-
[40]
Z. B. Maksic, D. Kovacek, M. Eckert-Maksic, M. Bockmann, M. Klessinger, Linear vs angular phenylenes: An interplay of aromaticity, antiaromaticity, and baeyer strain in fused molecular systems, The Journal of Physical Chemistry 99 (17) (1995) 6410–6416.arXiv:https://doi.org/10.1021/j100017a020,doi:10.1021/ j100017a020. URLhttps://doi.org/10.1021/j100017a020
-
[41]
Soetewey, Two-way ANOV A in R, accessed: 29 Aug 2025 (Jun
A. Soetewey, Two-way ANOV A in R, accessed: 29 Aug 2025 (Jun. 2023). URLhttps://statsandr.com/blog/two-way-anova-in-r/
work page 2025
-
[42]
N. J. Salkind, Encyclopedia of research design, SAGE, Los Angeles, [Calif.] ;, 2010. URLhttps://login.eux.idm.oclc.org/login?url=http://sk.sagepub.com/reference/ researchdesign
work page 2010
-
[43]
F. de Mendiburu, agricolae: Statistical Procedures for Agricultural Research, r package version 1.3-7 (2023). URLhttps://CRAN.R-project.org/package=agricolae
work page 2023
-
[44]
S. H. Yalkowsky, Carnelley’s rule and the prediction of melting point, Journal of Pharmaceutical Sciences 103 (9) (2014) 2629–2634.doi:https://doi.org/10.1002/jps.24034. URLhttps://www.sciencedirect.com/science/article/pii/S0022354915304445
-
[45]
D. Yoon, Y .-W. Son, H. Cheong, Negative thermal expansion coefficient of graphene measured by raman spec- troscopy, Nano letters 11 (8) (2011) 3227–3231.doi:10.1021/nl201488g. URLhttp://dx.doi.org/10.1021/nl201488g 13
-
[46]
M. Zahabul Islam, M. Mahboob, L. Robert Lowe, E. Stephen Bechtel, Characterization of the thermal expansion properties of graphene using molecular dynamics simulations, Journal of Physics D: Applied Physics 46 (43) (2013) 435302.doi:10.1088/0022-3727/46/43/435302. URLhttps://doi.org/10.1088/0022-3727/46/43/435302
-
[47]
Y . Magnin, G. D. Förster, F. Rabilloud, F. Calvo, A. Zappelli, C. Bichara, Thermal expansion of free-standing graphene: benchmarking semi-empirical potentials, Journal of Physics: Condensed Matter 26 (18) (2014) 185401.doi:10.1088/0953-8984/26/18/185401. URLhttps://doi.org/10.1088/0953-8984/26/18/185401
-
[48]
S. Mann, R. Kumar, V . K. Jindal, Negative thermal expansion of pure and doped graphene, RSC Adv. 7 (2017) 22378–22387.doi:10.1039/C7RA01591G. URLhttp://dx.doi.org/10.1039/C7RA01591G
-
[49]
Q. Feng, D. Wei, Y . Su, Z. Zhou, F. Wang, C. Tian, Study of thermal expansion coefficient of graphene via raman micro-spectroscopy: Revisited, Small 17 (12) (2021) 2006146.doi:10.1002/smll.202006146. URLhttps://doi.org/10.1002/smll.202006146
-
[50]
functionalized carbophenes as high-capacity versatile gas adsorbents: An ab initio study
C. E. Junkermeier, E. Larmand, J.-C. Morais, J. Kobebel, K. Lavarez, R. M. Adra, J. Yang, V . A. Diaz, R. Paupitz, G. Psofogiannakis, Corrigendum to “functionalized carbophenes as high-capacity versatile gas adsorbents: An ab initio study” [comput. mater. sci. 232 (2023) 112665], Computational Materials Science 239 (2024) 112921. doi:https://doi.org/10.10...
-
[51]
Q. Li, Y . Li, Y . Chen, L. Wu, C. Yang, X. Cui, Synthesis ofγ-graphyne by mechanochemistry and its electronic structure, Carbon 136 (2018) 248–254.doi:https://doi.org/10.1016/j.carbon.2018.04.081. URLhttps://www.sciencedirect.com/science/article/pii/S0008622318304421
-
[52]
N. Tyutyulkov, F. Dietz, K. Müllen, M. Baumgarten, Structure and energy spectra of a two-dimensional dielectric carbon allotrope, Chemical Physics Letters 272 (1) (1997) 111 – 114.doi:https://doi.org/10.1016/ S0009-2614(97)00465-X. URLhttp://www.sciencedirect.com/science/article/pii/S000926149700465X
work page 1997
-
[53]
Q. Fan, L. Yan, M. W. Tripp, O. Krej ˇcí, S. Dimosthenous, S. R. Kachel, M. Chen, A. S. Foster, U. Koert, P. Liljeroth, J. M. Gottfried, Biphenylene network: A nonbenzenoid carbon allotrope, Science 372 (6544) (2021) 852–856.arXiv:https://www.science.org/doi/pdf/10.1126/science.abg4509,doi:10. 1126/science.abg4509. URLhttps://www.science.org/doi/abs/10.11...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.