Atomistic Modeling of Methane and Carbon Dioxide Structure I Gas Hydrates Under Pressure: Guest Effects and Properties
Pith reviewed 2026-05-10 13:49 UTC · model grok-4.3
The pith
Carbon dioxide molecules rotate to align with hydrate cage faces under pressure, dispersing energy changes that methane cannot.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The DFT simulations of sI methane and carbon dioxide hydrates find that carbon dioxide molecules align parallel to the hexagonal faces of the large cages under pressure, independent of the functional. This rotational freedom lets carbon dioxide disperse changes in the energy landscape, producing distinct property differences from methane hydrates. The approach maps the pressure-enthalpy landscape, elastic stability, and marginal stability while confirming experimentally seen restrictions on guest rotations and new pressure behaviors under hydrostatic loading.
What carries the argument
The rotational freedom and pressure-induced alignment of guest molecules inside sI hydrate cages, which redistributes energy landscape changes differently for carbon dioxide than for methane in the DFT calculations.
If this is right
- Carbon dioxide hydrates remain mechanically stable at higher pressures than methane hydrates due to rotational dispersion of energy changes.
- Methane hydrates exhibit more rigid response to pressure, leading to different elastic moduli and volumes.
- Both functionals predict the same alignment trend for carbon dioxide, though revPBE gives larger equilibrium volumes.
- The models identify regions of marginal stability useful for predicting hydrate behavior in storage or separation applications.
- Guest rotation restrictions observed in experiments are reproduced by the simulations.
Where Pith is reading between the lines
- Mixed methane-carbon dioxide hydrates might show tunable stability by varying the guest ratio and pressure.
- The rotation effect could extend to other polar or linear guest molecules in similar cage structures.
- Pressure could be used as a control variable in hydrate-based processes to select between different gases based on their rotational response.
Load-bearing premise
The chosen DFT functionals and simulation cell sizes accurately capture the real intermolecular forces and long-range order in the hydrates without large functional or finite-size errors.
What would settle it
An experimental measurement such as neutron diffraction or Raman spectroscopy showing that carbon dioxide molecules do not align parallel to the hexagonal faces of large cages in sI hydrates under hydrostatic pressure.
Figures





read the original abstract
Gas hydrates are potential candidates in future energy sources while simultaneously providing structures with extensive applications in carbon capture and storage, gas transport, and important separation processes. Prior research in the field considers the dynamics of the water molecule backbone in particular. We investigated the pressure-enthalpy landscape and mechanical stability envelope of sI methane and carbon dioxide hydrates simulated using DFT. We investigated the effect of the revPBE + DFT-D2 and the SCAN + rVV10 and their treatment of the exchange correlation interactions. We examined the zero pressure material properties, finding that revPBE comparatively underbinds the interactions, causing more flexible structures with large equilibrium volumes. Under pressure, the carbon dioxide molecule was found to align itself parallel to the hexagonal faces of the large cage despite the functional used. Additionally, the property differences are caused by the ability of the carbon dioxide molecule to rotate and disperse the changes in the energy landscape in ways that methane molecules cannot. This computational methodology describes the elastic stability of gas hydrate, marginal stability, and critical differences across important molecular interactions, confirming experimentally observed restrictions in guest molecule rotations and novel pressure behaviors under hydrostatic loads
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript reports DFT-based atomistic simulations of structure I (sI) methane and carbon dioxide gas hydrates, focusing on their pressure-dependent enthalpy landscapes, elastic properties, and mechanical stability using the revPBE+DFT-D2 and SCAN+rVV10 functionals. Key findings include larger equilibrium volumes with revPBE due to underbinding, pressure-induced alignment of CO2 molecules parallel to hexagonal cage faces, and attribution of differences in properties between the two hydrates to the rotational freedom of CO2 allowing dispersion of energy landscape changes, unlike rigid CH4. The work claims to confirm experimental restrictions on guest rotations and novel pressure behaviors.
Significance. If the central interpretations hold, this study offers valuable computational insights into guest molecule effects on hydrate stability under pressure, relevant to carbon capture and energy applications. The direct use of DFT without empirical fitting parameters is a strength, providing a parameter-free approach to comparing functionals and guest behaviors. The identification of marginal stability and functional-dependent flexibility could inform models of hydrate behavior in geological or industrial settings. However, the significance is tempered by the need for stronger evidence on the proposed causal mechanism.
major comments (1)
- [Abstract] Abstract: the claim that 'the property differences are caused by the ability of the carbon dioxide molecule to rotate and disperse the changes in the energy landscape in ways that methane molecules cannot' is not supported by the reported results, which describe only static alignments under pressure and functional-dependent volumes. No rotational energy barriers, free-energy sampling, or rotation-suppressed control calculations are presented to establish the dispersion mechanism as the cause of elastic or enthalpic differences. This causal step is load-bearing for the guest-effects interpretation.
minor comments (3)
- Quantitative comparisons to experimental lattice parameters, bulk moduli, or stability limits are absent; the general claim of 'confirmation' would be strengthened by tabulated deviations or error estimates.
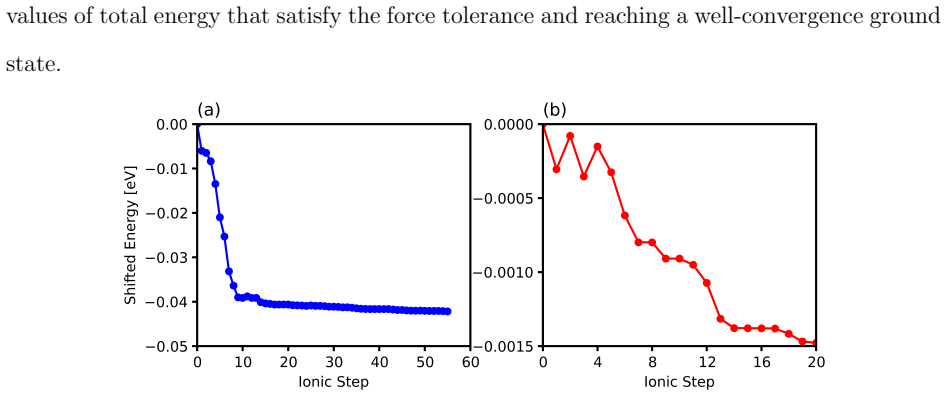
- Convergence with respect to supercell size, k-point density, and pressure increment should be documented to support the reported marginal stability and pressure behaviors.
- The specific figures or tables quantifying the CO2 alignment (e.g., orientation angles or energy differences) and the functional dependence of flexibility should be cross-referenced more explicitly in the text.
Simulated Author's Rebuttal
We thank the referee for their careful reading and constructive comments on our manuscript. The concern about the causal interpretation of guest effects is well-taken, and we have revised the abstract and discussion to align the claims more closely with the static DFT evidence while preserving the scientific interpretation supported by the structural results.
read point-by-point responses
-
Referee: [Abstract] Abstract: the claim that 'the property differences are caused by the ability of the carbon dioxide molecule to rotate and disperse the changes in the energy landscape in ways that methane molecules cannot' is not supported by the reported results, which describe only static alignments under pressure and functional-dependent volumes. No rotational energy barriers, free-energy sampling, or rotation-suppressed control calculations are presented to establish the dispersion mechanism as the cause of elastic or enthalpic differences. This causal step is load-bearing for the guest-effects interpretation.
Authors: We agree that the original abstract phrasing asserted a direct causal mechanism without explicit dynamical evidence such as rotational barriers or free-energy sampling. Our DFT optimizations at multiple pressures demonstrate that CO2 consistently adopts alignments parallel to the large-cage hexagonal faces, yielding multiple symmetry-equivalent low-energy orientations unavailable to rigid CH4. These orientational preferences are reproduced with both functionals and coincide with the observed differences in pressure-dependent enthalpies and elastic constants. We interpret the ability of CO2 to select among these orientations as enabling dispersion of lattice strain, but we acknowledge this remains an inference from static structures rather than a quantified dynamical process. In revision we have replaced the causal language in the abstract with a statement that the property differences are associated with the pressure-induced orientational freedom of CO2, as evidenced by the alignments. We have added a clarifying paragraph in the discussion noting that molecular-dynamics or free-energy calculations would be required to confirm the energetic dispersion mechanism and have referenced experimental studies on guest rotational restrictions in sI hydrates to support the broader interpretation. revision: yes
Circularity Check
No significant circularity; results from direct DFT simulation
full rationale
The paper reports DFT-based atomistic simulations of sI methane and CO2 hydrates using revPBE+DFT-D2 and SCAN+rVV10 functionals. Zero-pressure volumes, pressure-induced alignments, elastic stability, and guest-specific behaviors are computed directly from the electronic structure calculations. The attribution of property differences to CO2 rotational freedom is presented as an interpretation of observed static alignments and energy landscapes, not as a derived equation that reduces to a fitted parameter or prior self-citation. No self-definitional steps, fitted inputs renamed as predictions, or load-bearing self-citations appear in the abstract or described methodology. The chain remains self-contained, with results benchmarked against experimental rotation restrictions rather than internally forced.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Standard density functional theory approximations for exchange-correlation energy are sufficient to describe water-guest interactions in sI hydrates
Reference graph
Works this paper leans on
-
[1]
Scuseria, G. E.; Perdew, J. P. Density Functionals That Recognize Covalent, Metallic, and Weak Bonds.111, 106401. (36) Zhao, Y.; Truhlar, D. G. A New Local Density Functional for Main-Group Thermo- chemistry, Transition Metal Bonding, Thermochemical Kinetics, and Noncovalent In- teractions.125, 194101. (37) Sloan, E. D.; Koh, C. A. C. A.Clathrate Hydrates...
work page 1945
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.