Environment-dependent tight-binding models from ab initio pseudo-atomic orbital Hamiltonians
Pith reviewed 2026-05-10 12:25 UTC · model grok-4.3
The pith
Fitting environment-dependent tight-binding parameters simultaneously to pseudo-atomic orbital spectra from several atomic configurations produces transferable models with ab initio accuracy.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Ab initio pseudo-atomic orbital Hamiltonians already provide an exact representation of the electronic structure in a localized basis equivalent to a Slater-Koster tight-binding model. By augmenting the hopping integrals with bond-screening functions that capture the local environment and determining every parameter through a single fit to the PAO eigenvalue spectrum over several atomic configurations, the degeneracy between screening and hopping is broken. This produces physically meaningful, transferable models that generate Hamiltonians for large systems at ab initio precision, as shown for bulk platinum, silicon surfaces, Si/Ge superlattices, and twisted bilayer graphene containing up to
What carries the argument
bond-screening functions that augment Slater-Koster hopping integrals to encode local coordination environment
If this is right
- Hamiltonians for systems with thousands of atoms, such as twisted bilayer graphene, can be generated and post-processed for electronic, optical, and transport properties at ab initio accuracy.
- The same parameter set works across bulk metals, semiconductor surfaces, and heterostructures without re-fitting.
- The models integrate directly with existing post-processing suites to evaluate a wide range of properties on large-scale structures.
- All parameters acquire clear physical meaning once the screening-hopping degeneracy is lifted by multi-configuration fitting.
Where Pith is reading between the lines
- The approach could be applied to materials containing defects or interfaces by generating fitting configurations that include those features.
- Extension to time-dependent or non-equilibrium simulations becomes feasible once the efficient Hamiltonian generation is combined with molecular-dynamics drivers.
- The same fitting strategy might be used to incorporate additional interactions, such as spin-orbit terms, by including them in the underlying PAO reference calculations.
Load-bearing premise
Parameters obtained from simultaneous fitting to a finite collection of atomic configurations remain accurate and transferable to configurations and system sizes never included in the fit.
What would settle it
Compute the tight-binding eigenvalues or band-derived properties for a new twisted-bilayer geometry or defect configuration outside the fitting set and compare them directly with a full ab initio calculation; large systematic deviations would show the transferability claim fails.
Figures




read the original abstract
\textit{Ab initio} pseudo-atomic orbital (PAO) Hamiltonians express the electronic structure of a solid in a compact, localized basis that spans the same Hilbert space as a conventional Slater--Koster tight-binding model, thereby providing an exact \textit{ab initio} representation without any loss of accuracy. Building on this correspondence, we develop an environment-dependent tight-binding (EDTB) framework in which Slater--Koster hopping integrals are augmented with bond-screening functions that capture the local coordination environment. All parameters are determined by fitting to the PAO eigenvalue spectrum across multiple atomic configurations simultaneously, which breaks the degeneracy between screening and hopping parameters and yields physically meaningful, transferable models capable of generating Hamiltonians for large systems with \textit{ab initio} precision. We demonstrate the efficiency and accuracy of the approach on four prototypical systems: bulk platinum, silicon surfaces, Si/Ge~[001] superlattices, and twisted bilayer graphene with up to $4{,}324$ atoms. The method is implemented in the \paoflow{} code and integrates seamlessly with its full post-processing suite, enabling the evaluation of a broad range of electronic, optical, and transport properties.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript develops an environment-dependent tight-binding (EDTB) framework in which Slater-Koster hopping integrals are augmented by bond-screening functions whose parameters are obtained by simultaneous fitting to ab initio PAO eigenvalue spectra from multiple atomic configurations. The resulting models are asserted to be transferable and capable of generating Hamiltonians for large systems at ab initio precision. Demonstrations are provided for bulk platinum, silicon surfaces, Si/Ge [001] superlattices, and twisted bilayer graphene containing up to 4324 atoms; the method is implemented within the paoflow code for subsequent electronic-structure post-processing.
Significance. If the transferability claim is substantiated, the approach would supply a practical route to accurate, environment-dependent tight-binding Hamiltonians for systems whose size precludes direct ab initio treatment, while retaining compatibility with existing PAO-based workflows and post-processing tools. The simultaneous multi-configuration fit is a potentially useful technical step toward resolving screening-hopping ambiguities.
major comments (2)
- [Results: twisted bilayer graphene] The central claim that the fitted models achieve 'ab initio precision' for large systems rests on transferability to atomic arrangements outside the training set. In the twisted-bilayer-graphene demonstration, the 4324-atom moiré cell is treated, yet no quantitative comparison of the reconstructed Hamiltonian eigenvalues or matrix elements against direct PAO results (or any independent benchmark) is reported for that configuration or for continuously varying twist angles and registry shifts not included in the fit.
- [Methods: fitting strategy] The fitting procedure is described as simultaneously matching PAO eigenvalues across multiple configurations to break the screening/hopping degeneracy. However, the manuscript supplies neither the number and diversity of training configurations, the achieved root-mean-square fitting errors, nor any cross-validation on held-out structures, leaving the uniqueness and physical interpretability of the extracted parameters unquantified.
minor comments (2)
- [Methods] Notation for the bond-screening functions and the precise functional form of the environment dependence should be stated explicitly with an equation number in the methods section to allow readers to reproduce the model.
- [Figures] Figure captions for the TBG results should indicate the size of the training set relative to the 4324-atom cell and whether any test configurations were used.
Simulated Author's Rebuttal
We thank the referee for the constructive comments and positive overall assessment of the work. We address each major comment below, providing clarifications and committing to revisions where appropriate.
read point-by-point responses
-
Referee: [Results: twisted bilayer graphene] The central claim that the fitted models achieve 'ab initio precision' for large systems rests on transferability to atomic arrangements outside the training set. In the twisted-bilayer-graphene demonstration, the 4324-atom moiré cell is treated, yet no quantitative comparison of the reconstructed Hamiltonian eigenvalues or matrix elements against direct PAO results (or any independent benchmark) is reported for that configuration or for continuously varying twist angles and registry shifts not included in the fit.
Authors: We agree that direct quantitative validation against PAO eigenvalues for the 4324-atom cell would strengthen the transferability claim. However, performing a full PAO calculation for a system of this size is computationally prohibitive, which is precisely the regime the method targets. The manuscript instead demonstrates consistency with expected moiré physics (flat bands, Dirac features) derived from the reconstructed Hamiltonian. To address the concern, we will add in the revision quantitative comparisons of eigenvalues and matrix elements for smaller twist angles and registry shifts (where direct PAO benchmarks are feasible) as well as error metrics on held-out configurations. revision: partial
-
Referee: [Methods: fitting strategy] The fitting procedure is described as simultaneously matching PAO eigenvalues across multiple configurations to break the screening/hopping degeneracy. However, the manuscript supplies neither the number and diversity of training configurations, the achieved root-mean-square fitting errors, nor any cross-validation on held-out structures, leaving the uniqueness and physical interpretability of the extracted parameters unquantified.
Authors: We thank the referee for highlighting this omission. The revised manuscript will include a new subsection (or supplementary table) specifying the exact number and diversity of training configurations for each material, the root-mean-square eigenvalue fitting errors achieved, and cross-validation results on held-out atomic configurations. These additions will quantify the robustness, uniqueness, and physical interpretability of the extracted screening and hopping parameters. revision: yes
Circularity Check
No circularity: parameters fitted directly to external ab initio PAO spectra
full rationale
The paper's derivation proceeds by establishing a correspondence between ab initio PAO Hamiltonians and Slater-Koster tight-binding models, then augmenting the latter with bond-screening functions whose parameters are obtained via simultaneous fitting to eigenvalue spectra from multiple atomic configurations. This fitting uses independent external data as input and resolves parameter degeneracy through the multi-configuration constraint rather than by internal redefinition or self-reference. Transferability to large systems (e.g., 4324-atom twisted bilayer graphene) is asserted on the basis of explicit demonstrations, not by construction from the training data alone. The sole mention of the paoflow implementation is incidental and does not carry any load-bearing premise. No step reduces to a tautology, fitted input renamed as prediction, or self-citation chain.
Axiom & Free-Parameter Ledger
free parameters (1)
- bond-screening function parameters
axioms (1)
- domain assumption PAO Hamiltonians span the same Hilbert space as conventional Slater-Koster tight-binding models
Reference graph
Works this paper leans on
-
[1]
J. C. Slater and G. F. Koster, Simplified LCAO method for the periodic potential problem, Phys. Rev.94, 1498 (1954)
work page 1954
-
[2]
W. A. Harrison,Electronic Structure and the Properties of Solids(Freeman, San Francisco, 1980)
work page 1980
-
[3]
D. A. Papaconstantopoulos and M. J. Mehl, The Slater– Koster tight-binding method: a computationally efficient and accurate approach, J. Phys.: Condens. Matter15, R413 (2003)
work page 2003
-
[4]
M.BuongiornoNardelli, F.T.Cerasoli, M.Costa, S.Cur- tarolo, R. De Gennaro, M. Fornari, L. Liyanage, A. R. Supka, and H. Wang, Paoflow: A utility to construct and operate on ab initio hamiltonians from the projec- tions of electronic wavefunctions on atomic orbital bases, including characterization of topological materials, Com- putational Materials Scienc...
work page 2018
-
[5]
F.T.Cerasoli, A.R.Supka, A.Jayaraj, M.Costa, I.Siloi, J. Sławińska, S. Curtarolo, M. Fornari, D. Ceresoli, and M. Buongiorno Nardelli, Advanced modeling of materi- als with paoflow 2.0: New features and software design, Computational Materials Science200, 110828 (2021)
work page 2021
-
[6]
L. Goodwin, A. J. Skinner, and D. G. Pettifor, Gener- ating transferable tight-binding parameters: Application to silicon, Europhys. Lett.9, 701 (1989)
work page 1989
-
[7]
R. E. Cohen, M. J. Mehl, and D. A. Papaconstantopou- los, First-principles phonon dispersion curves for bcc transition metals, Phys. Rev. B50, 14694 (1994)
work page 1994
-
[8]
M. J. Mehl and D. A. Papaconstantopoulos, Applications of a tight-binding total-energy method for transition and noble metals, Phys. Rev. B54, 4519 (1996)
work page 1996
-
[9]
N. Bernstein, M. J. Mehl, D. A. Papaconstantopoulos, N. C. Papanicolaou, M. Z. Bazant, and E. Kaxiras, En- ergetic, vibrational, and electronic properties of silicon using a nonorthogonal tight-binding model, Phys. Rev. B62, 4477 (2000)
work page 2000
-
[10]
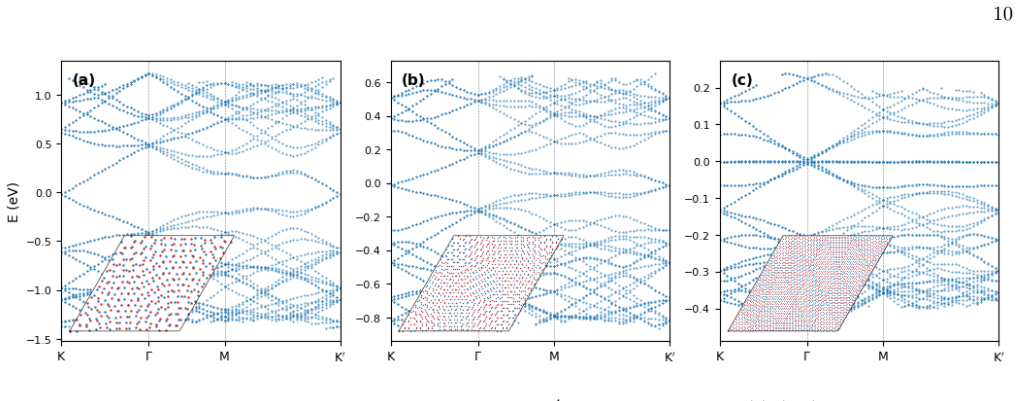
M. S. Tang, C. Z. Wang, C. T. Chan, and K. M. Ho, Environment-dependent tight-binding potential model, Phys. Rev. B53, 979 (1996). 10 FIG. 6. Sparse Lanczos band structure of TBG alongK–Γ–M–K′ for three supercells: (a)(6,5),θ= 6.01, 364 atoms; (b)(10,9),θ= 3.48, 1,084 atoms; (c)(20,19),θ= 1.70, 4,324 atoms. 40 eigenvalues nearestσ= 0.0eV are plotted perk-...
work page 1996
- [11]
-
[12]
L. A. Agapito, S. Ismail-Beigi, S. Curtarolo, M. Fornari, and M. Buongiorno Nardelli, Accurate tight-binding Hamiltonian matrices fromab initiocalculations: Mini- mal basis sets, Phys. Rev. B93, 035104 (2016)
work page 2016
-
[13]
L. A. Agapito, M. Fornari, D. Ceresoli, A. Ferretti, S. Curtarolo, and M. Buongiorno Nardelli, Accurate tight-binding hamiltonians for two-dimensional and lay- ered materials, Phys. Rev. B93, 125137 (2016)
work page 2016
-
[14]
P. D’Amico, L. Agapito, A. Catellani, A. Ruini, S. Cur- tarolo, M. Fornari, M. Buongiorno Nardelli, and A. Cal- zolari, Accurate ab initio tight-binding hamiltonians: Ef- fective tools for electronic transport and optical spec- troscopy from first principles, Phys. Rev. B94, 165166 (2016)
work page 2016
-
[15]
P. Giannozziet al., QUANTUM ESPRESSO: a modular and open-source software project for quantum simula- tions of materials, J. Phys.: Condens. Matter21, 395502 (2009)
work page 2009
-
[16]
Giannozziet al., Advanced capabilities for materials modelling with Quantum ESPRESSO, J
P. Giannozziet al., Advanced capabilities for materials modelling with Quantum ESPRESSO, J. Phys.: Con- dens. Matter29, 465901 (2017)
work page 2017
-
[17]
H.-J.Kim,TBFIT:Tight-bindingparameterfittingpack- age (2018)
work page 2018
-
[18]
M. Nakhaee, S. A. Ketabi, and F. M. Peeters, TB Studio: a technical tool for evaluating the electronic properties of solids, Comput. Phys. Commun.254, 107379 (2020)
work page 2020
-
[19]
G. Hegde, M. Povolotskyi, T. Kubis, T. B. Boykin, and G. Klimeck, An environment-dependent semi-empirical tight binding model suitable for electron transport in bulk metals, metal alloys, metallic interfaces, and metal- lic nanostructures. I. Model and validation, J. Appl. Phys.115, 123703 (2014)
work page 2014
-
[20]
Levenberg, A method for the solution of certain non- linear problems in least squares, Q
K. Levenberg, A method for the solution of certain non- linear problems in least squares, Q. Appl. Math.2, 164 (1944)
work page 1944
-
[21]
D. W. Marquardt, An algorithm for least-squares estima- tionofnonlinearparameters,J.Soc.Ind.Appl.Math.11, 431 (1963)
work page 1963
-
[22]
G. Hegde,Generation and optimization of tight binding (TB) parameters using genetic algorithms and their vali- dation using NEMO3D, Ph.D. thesis, Purdue University (2010)
work page 2010
-
[23]
A.-L. Phan, D. Soccodato, A. Pecchia, A. Di Vito, and M. Auf der Maur, Obtaining highly transferable param- eters for empirical tight-binding simulations: From pure materials to polytypic heterostructures and dilute alloys, Phys. Rev. B112, 165202 (2025)
work page 2025
-
[24]
A. G. Frøseth, R. Holmestad, P. M. Derlet, and K. Marthinsen, Improved tight-binding parametrization for the simulation of stacking faults in aluminum, Phys. Rev. B68, 012105 (2003)
work page 2003
-
[25]
K. F. Garrity and K. Choudhary, Fast and accurate prediction of material properties with three-body tight- binding model for the periodic table, Phys. Rev. Mater. 7, 044603 (2023)
work page 2023
-
[26]
Z. Wang, S. Ye, H. Wang, J. He, Q. Huang, and S. Chang, Machine learning method for tight-binding Hamiltonian parameterization fromab initioband struc- ture, npj Comput. Mater.7, 11 (2021)
work page 2021
-
[27]
S. Kim, Construction of optimized tight-binding mod- els using ab initio hamiltonian: application to monolayer 2h-transition metal dichalcogenides, Journal of Physics: Condensed Matter35, 415501 (2023)
work page 2023
-
[28]
PAOFLOW development team, In preparation (2026), in preparation
work page 2026
-
[29]
G. Y. Guo, Y. Yao, and Q. Niu,Ab initiocalculation of the intrinsic spin Hall effect in semiconductors, Phys. Rev. Lett.94, 226601 (2005)
work page 2005
-
[30]
G. Y. Guo, S. Murakami, T.-W. Chen, and N. Nagaosa, Intrinsic spin Hall effect in platinum: First-principles cal- culations, Phys. Rev. Lett.100, 096401 (2008)
work page 2008
-
[31]
J. Qiao, J. Zhou, Z. Yuan, and W. Zhao, Calculation of intrinsic spin Hall conductivity by Wannier interpolation, Phys. Rev. B98, 214402 (2018)
work page 2018
-
[32]
T. B. Boykin, G. Klimeck, M. A. Eriksson, M. Friesen, S. N. Coppersmith, P. von Allmen, F. Oyafuso, and S. Lee, Valley splitting in strained silicon quantum wells, Phys. Rev. B70, 165325 (2004)
work page 2004
-
[33]
M. Friesen, S. Chutia, C. Tahan, and S. N. Coppersmith, Valley splitting theory of SiGe/Si/SiGe quantum wells, 11 Phys. Rev. B75, 115318 (2007)
work page 2007
-
[34]
C. Lanczos, An iteration method for the solution of the eigenvalue problem of linear differential and integral op- erators, J. Res. Nat. Bur. Standards45, 255 (1950)
work page 1950
-
[35]
D. C. Sorensen, Implicit application of polynomial filters in ak-step Arnoldi method, SIAM J. Matrix Anal. Appl. 13, 357 (1992)
work page 1992
-
[36]
R. Bistritzer and A. H. MacDonald, Moiré bands in twisted double-layer graphene, Proc. Natl. Acad. Sci. USA108, 12233 (2011)
work page 2011
-
[37]
Anthropic, Introducing Claude 4 (2025), accessed: 2026- 04-04
work page 2025
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.