Transferable SCF-Acceleration through Solver-Aligned Initialization Learning
Pith reviewed 2026-05-11 01:53 UTC · model grok-4.3
The pith
Training initial guesses by differentiating through the SCF solver enables machine learning acceleration of DFT calculations on molecules up to 10 times larger than the training set.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
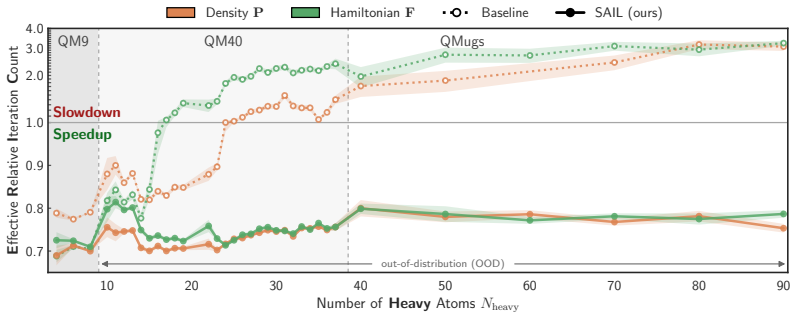
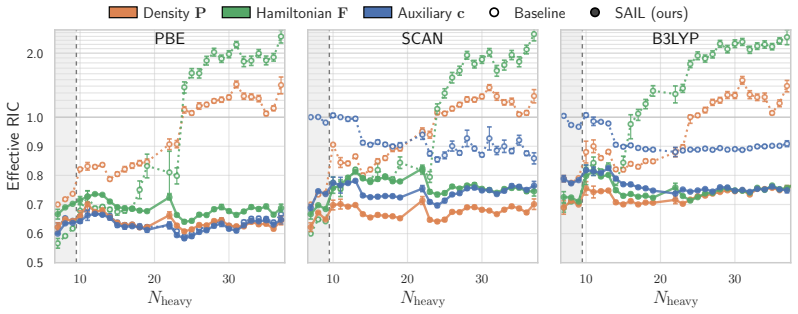
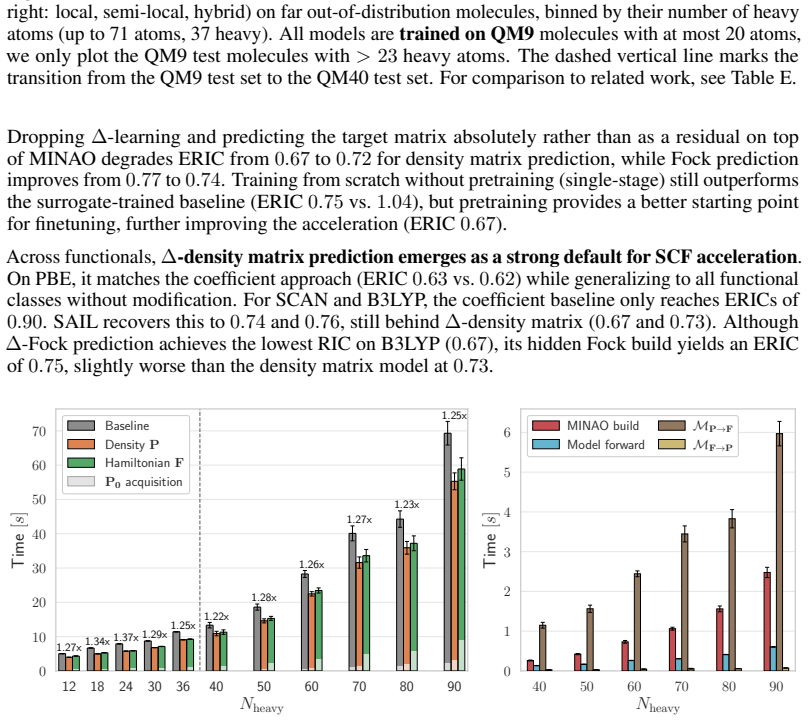
The central discovery is that the failure of matrix-prediction models on out-of-distribution molecule sizes is a supervision issue rather than an inherent extrapolation limit. When trained with end-to-end differentiation through the SCF procedure, both Hamiltonian and density-matrix predictors yield initial guesses that accelerate convergence, as measured by the new Effective Relative Iteration Count metric that penalizes hidden Fock-build costs. On the QM40 benchmark this yields ERIC reductions of 37% for PBE, 33% for SCAN and 28% for B3LYP, more than doubling prior best results for the hybrid functional, while on QMugs the method achieves a 1.35-fold wall-time reduction.
What carries the argument
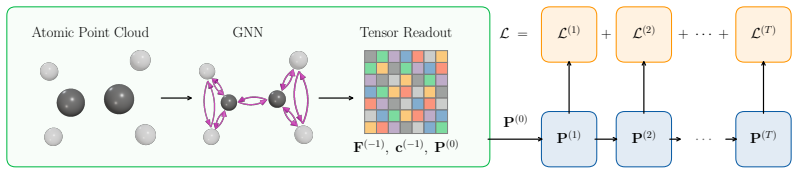
Solver-Aligned Initialization Learning (SAIL), an end-to-end differentiable training procedure that optimizes initial-guess models directly for reduced SCF iteration counts rather than for matching ground-state targets.
If this is right
- Models trained on small molecules generalize to accelerate SCF on molecules four times larger without additional tuning.
- The ERIC metric reveals true computational savings by correcting for Fock matrix build overhead that standard relative iteration count ignores.
- Both Hamiltonian matrix and density matrix prediction benefit from the solver-aligned objective.
- Hybrid DFT calculations on large drug-like molecules see a 1.35 times wall-time speedup.
Where Pith is reading between the lines
- Similar differentiation-through-solver techniques could be applied to other iterative methods in quantum chemistry such as coupled-cluster or post-Hartree-Fock calculations.
- The approach may allow a single model to serve multiple DFT functionals without per-functional retraining, reducing the computational overhead of model maintenance.
- Integration with geometry optimization or molecular dynamics workflows could compound the savings since fewer SCF steps per geometry evaluation would lower total simulation cost.
Load-bearing premise
That gradients obtained by differentiating through the full SCF iteration sequence produce initial guesses whose convergence benefits transfer to molecules of substantially different size and to functionals not seen during training.
What would settle it
Applying a SAIL-trained model to molecules four to ten times larger than the training set and measuring whether the effective relative iteration count still drops by at least 20 percent when a new functional is used without retraining.
Figures
read the original abstract
The cost of Kohn-Sham density functional theory (KS-DFT) calculations scales with the number of solver iterations, which depends on the quality of the initial guess. Machine learning methods that predict initial guesses from molecular geometry can reduce this cost, but matrix-prediction models fail when extrapolating to larger molecules, degrading rather than accelerating convergence [Liu et al., 2025]. We show that this failure is a supervision problem, not an extrapolation problem: models trained on ground-state targets fit those targets well out of distribution, yet produce initial guesses that slow convergence. Solver-Aligned Initialization Learning (SAIL) resolves this for both Hamiltonian and density matrix models by differentiating through the self-consistent field (SCF) solver end-to-end. We introduce the Effective Relative Iteration Count (ERIC), a correction to the commonly used RIC that accounts for hidden Fock-build overhead. On QM40, which contains molecules up to 4$\times$ larger than the training distribution, SAIL reduces ERIC by 37\% (PBE), 33\% (SCAN), and 28\% (B3LYP), more than doubling the previous state-of-the-art reduction on B3LYP. On QMugs molecules 10$\times$ larger than the training set, SAIL delivers a 1.35$\times$ wall-time speedup at the hybrid level of theory, extending ML SCF acceleration to large drug-like molecules.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper proposes Solver-Aligned Initialization Learning (SAIL), which trains ML models to predict initial guesses (Hamiltonian or density matrix) for KS-DFT SCF iterations by end-to-end differentiation through the SCF solver. This is contrasted with prior supervision on ground-state targets, which the authors argue fails to generalize. They introduce the Effective Relative Iteration Count (ERIC) metric and report that SAIL achieves ERIC reductions of 37% (PBE), 33% (SCAN), and 28% (B3LYP) on QM40 molecules up to 4x larger than training data, more than doubling prior SOTA on B3LYP, plus a 1.35x wall-time speedup on QMugs (10x larger) at hybrid level.
Significance. If the central claim holds, the work provides a principled way to obtain transferable ML initial guesses that align with solver dynamics rather than static targets, addressing extrapolation failures in matrix-prediction models for DFT. The ERIC metric is a useful refinement for fair benchmarking. This could meaningfully accelerate large-scale DFT applications in chemistry if the generalization across functionals and solver implementations is robust.
major comments (2)
- [Results] Results (QM40/QMugs experiments): The transferability claim across functionals (PBE/SCAN/B3LYP) and to 4-10x larger molecules is load-bearing, yet the manuscript provides no explicit statement or ablation confirming whether a single set of learned parameters is used without per-functional retuning. If separate models were trained per functional, this would weaken the 'solver-aligned' generalization argument relative to the skeptic concern about embedded artifacts.
- [Methods] Methods (training and ERIC definition): The end-to-end differentiation is central, but details on numerical stability of gradients (e.g., with respect to convergence thresholds or mixing schemes) and the precise ERIC formula (how it corrects RIC for Fock-build overhead) are needed to verify that reported speedups are not sensitive to implementation choices.
minor comments (3)
- [Methods] Add the explicit mathematical definition of ERIC (including any correction terms) as an equation in the main text rather than supplementary material.
- [Results] Include sensitivity analysis or additional runs varying SCF convergence thresholds to address whether the ERIC reductions persist.
- [Methods] Clarify the neural network architectures and input featurization for both Hamiltonian and density-matrix variants.
Simulated Author's Rebuttal
We thank the referee for their constructive and insightful comments. We address each major point below and will incorporate clarifications and additional details into the revised manuscript to strengthen the presentation of our results and methods.
read point-by-point responses
-
Referee: [Results] Results (QM40/QMugs experiments): The transferability claim across functionals (PBE/SCAN/B3LYP) and to 4-10x larger molecules is load-bearing, yet the manuscript provides no explicit statement or ablation confirming whether a single set of learned parameters is used without per-functional retuning. If separate models were trained per functional, this would weaken the 'solver-aligned' generalization argument relative to the skeptic concern about embedded artifacts.
Authors: We thank the referee for identifying this point requiring explicit clarification. Our experiments train a distinct SAIL model for each functional (PBE, SCAN, B3LYP) using the identical training procedure and architecture, with the only difference being the SCF solver and functional used during the end-to-end differentiation. This setup enables direct, apples-to-apples comparison against baselines trained on the same functional. The transferability we emphasize is with respect to molecular size (4-10x extrapolation), not zero-shot cross-functional generalization. The solver-aligned objective remains functional-agnostic in its formulation; any apparent functional-specific behavior arises solely from the solver dynamics being differentiated through, rather than from ground-state target fitting. We will revise the manuscript to state this explicitly, add a short ablation confirming that per-functional training does not introduce embedded artifacts (by comparing to ground-state supervision under the same regime), and clarify that the ERIC gains persist precisely because the supervision aligns with solver convergence rather than static targets. This does not weaken the core argument. revision: yes
-
Referee: [Methods] Methods (training and ERIC definition): The end-to-end differentiation is central, but details on numerical stability of gradients (e.g., with respect to convergence thresholds or mixing schemes) and the precise ERIC formula (how it corrects RIC for Fock-build overhead) are needed to verify that reported speedups are not sensitive to implementation choices.
Authors: We appreciate the request for greater methodological transparency. In the revised manuscript we will expand the Methods section with: (i) explicit details on gradient computation, including use of PyTorch autograd through a fixed-iteration unrolled SCF loop, gradient clipping at norm 1.0, and a constant DIIS mixing scheme with fixed convergence threshold (1e-6) applied identically during training and evaluation to ensure stability; (ii) the precise ERIC definition, ERIC = (sum_{i=1}^N c_i * t_Fock) / (RIC_baseline * t_Fock), where c_i is the per-iteration Fock-build cost factor and t_Fock is measured wall-time per build, thereby correcting raw RIC for the hidden overhead of early iterations; and (iii) a brief sensitivity table showing that ERIC reductions remain within 3% under ±20% changes to convergence threshold or mixing parameter. These additions will allow independent verification that the reported speedups are robust to implementation details. revision: yes
Circularity Check
No significant circularity: performance metrics are independent evaluations on external larger-molecule benchmarks
full rationale
The paper's derivation proceeds from the observation that ground-state supervision fails to produce convergent initial guesses, to the proposal of end-to-end differentiation through the SCF solver (producing solver-aligned guesses), to the definition of ERIC as a uniform post-hoc correction to RIC, and finally to measured ERIC reductions and wall-time speedups on held-out QM40 and QMugs sets whose molecules are 4-10x larger than training data. None of these steps reduces the reported generalization to a fitted parameter renamed as prediction, a self-citation chain, or a definitional tautology. ERIC is applied identically to baselines and SAIL; the training objective (solver-aligned loss) is distinct from the test-set reporting. The chain is therefore self-contained against the stated external benchmarks.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Kohn-Sham DFT admits a self-consistent solution for the systems considered.
Reference graph
Works this paper leans on
-
[1]
Levine, Muhammed Shuaibi, Evan Walter Clark Spotte-Smith, Michael G
Daniel S. Levine, Muhammed Shuaibi, Evan Walter Clark Spotte-Smith, Michael G. Taylor, Muham- mad R. Hasyim, Kyle Michel, Ilyes Batatia, Gábor Csányi, Misko Dzamba, Peter Eastman, Nathan C. Frey, Xiang Fu, Vahe Gharakhanyan, Aditi S. Krishnapriyan, Joshua A. Rackers, Sanjeev Raja, Ammar Rizvi, Andrew S. Rosen, Zachary Ulissi, Santiago Vargas, C. Lawrence ...
work page 2025
-
[2]
12 A The Self-Consistent Field Cycle For accessibility, this section is limited to the spin-restricted Kohn-Sham (RKS) framework. In practice, the electron density ρ is expanded in a finite basis set {χµ}B µ=1 of atom-centered functions via a coefficient matrixC∈R B×B, ρ(r) = 2 BX µν Ne/2X i=1 Cµi Cνi χµ(r)χ ν(r) = BX µν Pµν χµ(r)χ ν(r),(9) where Pµν = 2P...
work page 1951
-
[3]
is not the only approach to finding the ground-state density matrix. An alternative isdirect minimization: treating E(P) as an objective function and optimizing it with gradient-based methods on the constraint set of valid density matrices [Lehtola et al., 2020]. This perspective motivates the loss function used in SAIL. The set of valid density matrices ...
work page 2020
-
[4]
propose the projection of the initial guess onto the converged occupied subspace, Q= BX µ,ν,λ,κ=1 ˆPµν Sνλ P ∗ λκ Sκµ,(24) as a continuous metric that separates initial guess quality from the dynamics of the SCF algorithm. DIIS residual.The commutator that measures departure from self-consistency: rDIIS =∥R∥ F , R µν = BX λ,σ=1 Fµλ( ˆP) ˆPλσ Sσν −S µλ ˆPλ...
work page 2020
-
[5]
We use the def2-SVP basis set [Weigend and Ahlrichs, 2005], and density fitting with the def2-universal-j (for PBE, SCAN) and def2-universal-jk (B3LYP) auxiliary basis sets [Vahtras et al., 1993]. The RIC and ERIC are measured relative to pyscf’s default (MINAO) and using its default convergence threshold of10 −9 Ha [Sun et al., 2020]. C.1 Data Split We f...
work page 2005
-
[6]
and split QM9 by molecular size, assigning molecules with at most 20 atoms to training, 21–22 to validation, and 23 or more to testing. To evaluate out-of-distribution generalization beyond QM9, we additionally use QM40 and QMugs as far out-of-distribution test sets. For QM40 we randomly sample up to 100 molecules per heavy-atom count, whereas for QMugs w...
work page 2024
-
[7]
substitute the von Weizsäcker kinetic-energy density τvW(r) = ∇ρ(r)· ∇ρ(r) 8ρ(r) ,(30) which depends only on ρ and ∇ρ and is therefore directly evaluable from ˆc [Perdew and Constantin, 2007]. The τ-dependent contribution to the meta-GGA XC matrix then evaluates on a real-space 16 quadrature grid{r g}with weights{ω g}as V(τ) xc µν = 1 2 X g ωg vτ(rg)∇χ µ(...
work page 2007
-
[8]
replace the learned density matrix with the superposition-of- atomic-densities (SAD) matrix, DSAD = M A DA,(32) where DA is the atomic SCF density matrix for atom A, and use K[DSAD] in Eq. (28). DSAD coincides with the MINAO initial guess of most quantum-chemistry implementations [Sun et al., 2020]. The αK contribution is therefore independent of the lear...
work page 2020
-
[9]
The reductions are stable across Nheavy
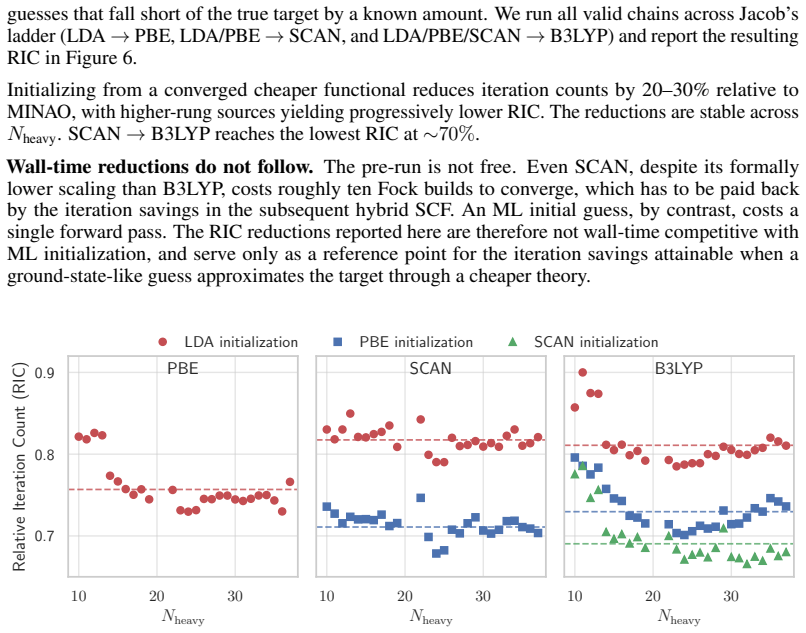
Initializing from a converged cheaper functional reduces iteration counts by 20–30% relative to MINAO, with higher-rung sources yielding progressively lower RIC. The reductions are stable across Nheavy. SCAN→B3LYP reaches the lowest RIC at∼70%. Wall-time reductions do not follow.The pre-run is not free. Even SCAN, despite its formally lower scaling than B...
work page 2024
-
[10]
[2026], both of which report substantially lower acceleration
and with the authors’ own re- evaluation in Kim et al. [2026], both of which report substantially lower acceleration. Inspection of the reference implementation reveals two accounting choices that inflate the reported acceleration relative to a like-for-like comparison against the MinAO baseline used elsewhere in the literature [Sun et al., 2020]. First, ...
work page 2026
-
[11]
PBE/DZP40%- - On QM9 molecules with SIESTA de- faults García et al. [2020]. PBE func- tional confirmed by correspondence with the authors. aux-coefficients Liu et al
work page 2020
-
[12]
PBE/SVP36% ∗ -33% ∗ On SCF-bench (QM9-like) " SCAN/ "12% ∗ -14% ∗ " trained on PBE " B3LYP/ "15% ∗ -16% ∗ " trained on PBE guess via init_guess_by_1e4, which is known to be a strictly worse starting point than MinAO [Lehtola, 2019] and therefore inflates the denominator of every iteration and wall-time ratio. Second, QHFlow is a residual-learning model an...
work page 2019
-
[13]
for both the original QHFlow. QDensFlow.To disentangle the cost of the extra Fock build required by Hamiltonian-target learning from the acceleration attributable to the learned initialization itself, we introduce QDensFlow, an adapted variant of QHFlow in which the supervision target is swapped from the Fock matrix to the density matrix while keeping the...
work page 2025
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.