Evaluating Post-hoc Explanations of the Transformer-based Genome Language Model DNABERT-2
Pith reviewed 2026-05-09 22:26 UTC · model grok-4.3
The pith
AttnLRP applied to DNABERT-2 produces explanations that align with known biological patterns in DNA sequences.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
The central claim is that AttnLRP yields reliable explanations for DNABERT-2 that correspond to known biological patterns, allowing genome language models to help derive biological insights similar to how convolutional neural networks do.
What carries the argument
AttnLRP, an extension of layer-wise relevance propagation adapted to handle the attention mechanisms in transformer models like DNABERT-2, which attributes relevance scores back to individual nucleotides or tokens in the input genome sequence.
If this is right
- Explanations can be transferred from token level to nucleotide level for direct biological interpretation.
- Genome language models produce explanations comparable to those from convolutional networks.
- Post-hoc methods like AttnLRP enable hypothesis generation from gLM predictions on genome sequences.
- Relevance attributions become comparable across transformer and convolutional architectures.
- gLMs can support biological insight extraction in the same way CNNs have been shown to do.
Where Pith is reading between the lines
- Similar adaptation of relevance methods could be tested on other genome language models to check consistency.
- Biologists could use the highlighted patterns to select candidate regions for targeted experiments.
- The approach might extend to non-genomic sequence tasks where transformers are applied.
- Performance gains from more complex models may not come at the cost of lost interpretability.
Load-bearing premise
The chosen evaluation metrics and comparison to a CNN baseline are sufficient to show that the explanations capture genuine biological relevance rather than model-specific artifacts or biases.
What would settle it
If AttnLRP explanations on a held-out genomic dataset with verified known motifs fail to highlight those motifs while the model still predicts accurately, that would show the attributions do not reflect biological patterns.
Figures
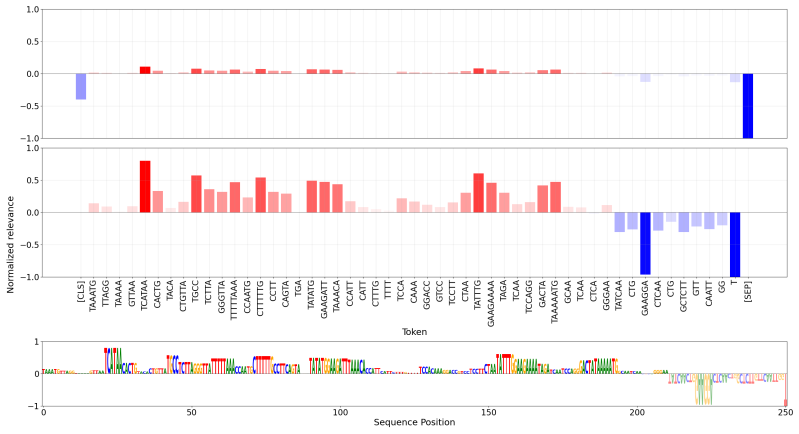
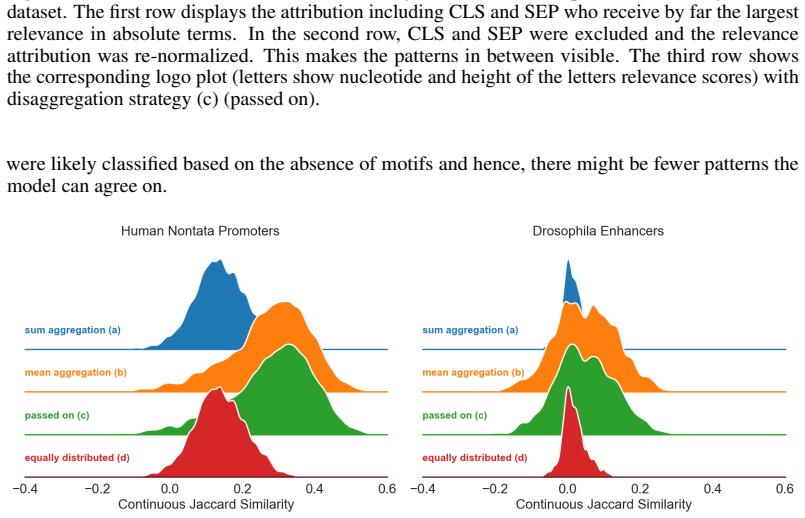
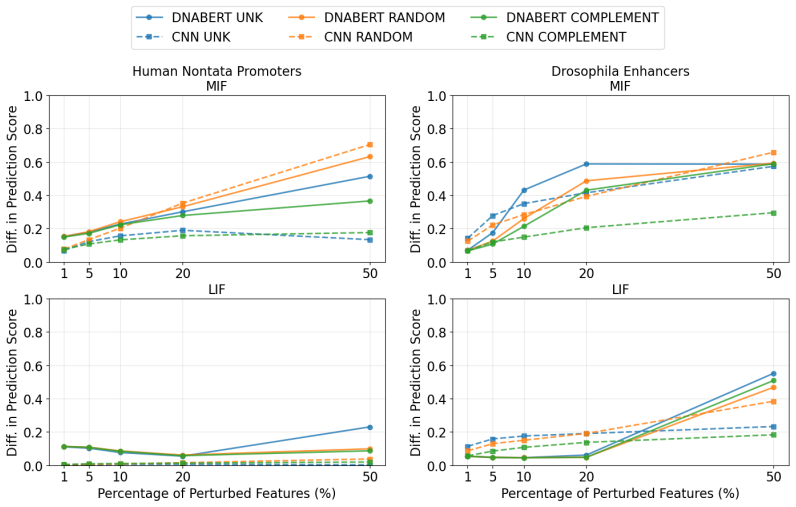
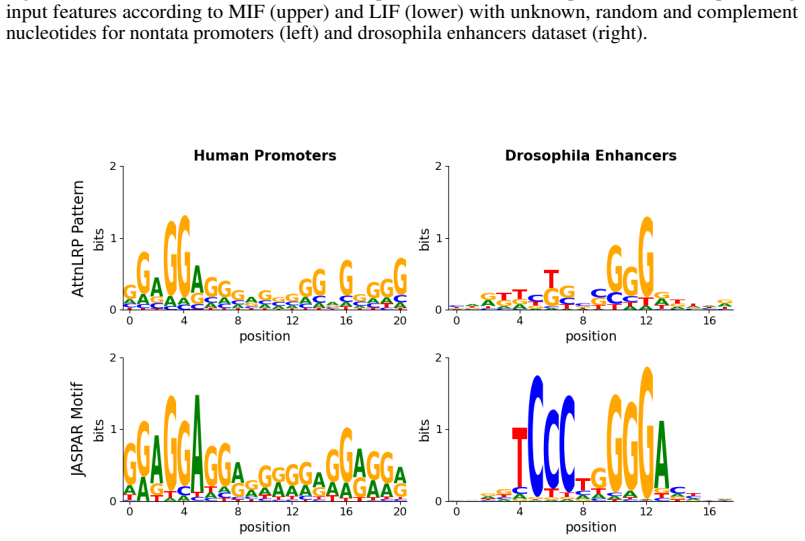
read the original abstract
Explaining deep neural network predictions on genome sequences enables biological insight and hypothesis generation-often of greater interest than predictive performance alone. While explanations of convolutional neural networks (CNNs) have been shown to capture relevant patterns in genome sequences, it is unclear whether this transfers to more expressive Transformer-based genome language models (gLMs). To answer this question, we adapt AttnLRP, an extension of layer-wise relevance propagation to the attention mechanism, and apply it to the state-of-the-art gLM DNABERT-2. Thereby, we propose strategies to transfer explanations from token and nucleotide level. We evaluate the adaption of AttnLRP on genomic datasets using multiple metrics. Further, we provide an extensive comparison between the explanations of DNABERT-2 and a baseline CNN. Our results demonstrate that AttnLRP yields reliable explanations corresponding to known biological patterns. Hence, like CNNs, gLMs can also help derive biological insights. This work contributes to the explainability of gLMs and addresses the comparability of relevance attributions across different architectures.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript adapts AttnLRP (an extension of layer-wise relevance propagation to attention) to the Transformer-based genome language model DNABERT-2. It introduces strategies for transferring token-level attributions to the nucleotide level and evaluates the resulting explanations on genomic datasets via multiple metrics (motif overlap, conservation, etc.), with an extensive comparison to a CNN baseline. The central claim is that AttnLRP produces reliable explanations that correspond to known biological patterns, implying that gLMs can derive biological insights in the same manner as CNNs.
Significance. If the evaluation holds after addressing controls, the work would meaningfully extend post-hoc explainability from CNNs to more expressive Transformer gLMs, supporting their use for hypothesis generation in genomics. The architecture-comparison angle and the explicit transfer strategies from token to nucleotide level are constructive contributions.
major comments (2)
- [Evaluation / Results] The evaluation lacks composition-matched negative controls (e.g., dinucleotide-frequency or GC-content shuffled sequences) that would distinguish attributions reflecting genuine functional elements from those driven by shared statistical biases between DNABERT-2 and the CNN baseline. This directly undermines the claim that the observed correspondence to biological patterns demonstrates reliable, biologically meaningful explanations rather than metric artifacts.
- [Evaluation / Results] No details are provided on the exact genomic datasets, precise definitions of the reported metrics (overlap, conservation scores, etc.), or statistical procedures (multiple-testing correction, permutation baselines). Without these, it is impossible to assess whether the positive results across metrics are robust or subject to post-hoc selection.
minor comments (2)
- [Abstract] The abstract states that explanations 'correspond to known biological patterns' but does not name the specific patterns or datasets; adding one concrete example (e.g., a particular motif or conservation track) would improve clarity.
- [Methods] Notation for the token-to-nucleotide transfer strategy should be formalized (e.g., an equation or pseudocode block) rather than described only in prose.
Simulated Author's Rebuttal
We thank the referee for the constructive and detailed comments, which highlight important aspects for strengthening the evaluation. We address each major comment below and indicate the corresponding revisions to the manuscript.
read point-by-point responses
-
Referee: The evaluation lacks composition-matched negative controls (e.g., dinucleotide-frequency or GC-content shuffled sequences) that would distinguish attributions reflecting genuine functional elements from those driven by shared statistical biases between DNABERT-2 and the CNN baseline. This directly undermines the claim that the observed correspondence to biological patterns demonstrates reliable, biologically meaningful explanations rather than metric artifacts.
Authors: We agree that composition-matched negative controls are a valuable addition to distinguish genuine biological signals from potential statistical artifacts shared across models. Although the CNN baseline comparison provides partial mitigation by holding the training data constant, it does not fully address compositional biases. In the revised manuscript we will incorporate dinucleotide-frequency and GC-content shuffled sequence controls, recompute the attribution metrics on these controls, and report the resulting reductions in motif overlap and conservation scores to demonstrate that the positive results are not artifacts. revision: yes
-
Referee: No details are provided on the exact genomic datasets, precise definitions of the reported metrics (overlap, conservation scores, etc.), or statistical procedures (multiple-testing correction, permutation baselines). Without these, it is impossible to assess whether the positive results across metrics are robust or subject to post-hoc selection.
Authors: We acknowledge that the original manuscript omitted sufficient detail on these elements. The revised version will add a dedicated subsection in the Methods section specifying the exact genomic datasets (including accession numbers, sequence lengths, and preprocessing), precise definitions of each metric (e.g., how motif overlap is quantified via position-specific scoring and how conservation scores are obtained from phastCons or equivalent tracks), and the complete statistical procedures (including multiple-testing corrections and permutation-based baselines for significance assessment). revision: yes
Circularity Check
No significant circularity in evaluation methodology
full rationale
The paper conducts an empirical evaluation of adapted AttnLRP explanations on DNABERT-2 by measuring overlap with known biological motifs, conservation scores, and comparison against a separate CNN baseline using multiple independent metrics. These steps rely on external genomic annotations and cross-architecture benchmarks rather than any self-referential definitions, fitted parameters renamed as predictions, or load-bearing self-citations that reduce the central claim to the paper's own inputs by construction. The derivation chain is self-contained against external data and does not exhibit any of the enumerated circular patterns.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
In: International Conference on Machine Learning
Achtibat, R., Hatefi, S.M.V ., Dreyer, M., Jain, A., Wiegand, T., Lapuschkin, S., Samek, W.: At- tnLRP: Attention-Aware Layer-Wise Relevance Propagation for Transformers. In: International Conference on Machine Learning. pp. 135–168. PMLR (2024)
work page 2024
-
[2]
In: International Conference on Machine Learning
Ali, A., Schnake, T., Eberle, O., Montavon, G., Müller, K.R., Wolf, L.: Xai for transformers: Better explanations through conservative propagation. In: International Conference on Machine Learning. pp. 435–451. PMLR (2022)
work page 2022
-
[3]
Nature genetics54(5), 613–624 (2022) 9
de Almeida, B.P., Reiter, F., Pagani, M., Stark, A.: DeepSTARR predicts enhancer activity from DNA sequence and enables the de novo design of synthetic enhancers. Nature genetics54(5), 613–624 (2022) 9
work page 2022
-
[4]
A close look at decomposition-based xai-methods for transformer language models, 2025
Arras, L., Puri, B., Kahardipraja, P., Lapuschkin, S., Samek, W.: A Close Look at Decomposition-based XAI-Methods for Transformer Language Models. arXiv preprint arXiv:2502.15886 (2025)
-
[5]
Bach, S., Binder, A., Montavon, G., Klauschen, F., Müller, K.R., Samek, W.: On Pixel-Wise Explanations for Non-Linear Classifier Decisions by Layer-Wise Relevance Propagation. PLoS ONE10(2015)
work page 2015
-
[6]
NAR genomics and bioinformatics3(1), lqab004 (2021)
Bartoszewicz, J.M., Seidel, A., Renard, B.Y .: Interpretable detection of novel human viruses from genome sequencing data. NAR genomics and bioinformatics3(1), lqab004 (2021)
work page 2021
-
[7]
Evaluating and aggregating feature-based model explanations,
Bhatt, U., Weller, A., Moura, J.M.: Evaluating and aggregating feature-based model explana- tions. arXiv preprint arXiv:2005.00631 (2020)
-
[8]
In: International Conference on Machine Learning
Chalasani, P., Chen, J., Chowdhury, A.R., Wu, X., Jha, S.: Concise explanations of neural networks using adversarial training. In: International Conference on Machine Learning. pp. 1383–1391. PMLR (2020)
work page 2020
-
[9]
In: Proceedings of the IEEE/CVF conference on computer vision and pattern recognition
Chefer, H., Gur, S., Wolf, L.: Transformer interpretability beyond attention visualization. In: Proceedings of the IEEE/CVF conference on computer vision and pattern recognition. pp. 782–791 (2021)
work page 2021
-
[10]
Briefings in bioinformatics22(5), bbab060 (2021)
Clauwaert, J., Menschaert, G., Waegeman, W.: Explainability in transformer models for functional genomics. Briefings in bioinformatics22(5), bbab060 (2021)
work page 2021
-
[11]
Nature Machine Intelligence pp
Consens, M.E., Dufault, C., Wainberg, M., Forster, D., Karimzadeh, M., Goodarzi, H., Theis, F.J., Moses, A., Wang, B.: Transformers and genome language models. Nature Machine Intelligence pp. 1–17 (2025)
work page 2025
-
[12]
In: 2023 ICML Workshop on Computational Biology, Honolulu, Hawaii, USA (2023)
Consens, M.E., Papernot, N., Wang, B., Moses, A.: Transforming Genomic Interpretability: A DNABERT Case Study. In: 2023 ICML Workshop on Computational Biology, Honolulu, Hawaii, USA (2023)
work page 2023
-
[13]
Nature Methods22(2), 287–297 (2025)
Dalla-Torre, H., Gonzalez, L., Mendoza-Revilla, J., Lopez Carranza, N., Grzywaczewski, A.H., Oteri, F., Dallago, C., Trop, E., de Almeida, B.P., Sirelkhatim, H., Richard, G., Skwark, M., Beguir, K., Lopez, M., Pierrot, T.: Nucleotide Transformer: building and evaluating robust foundation models for human genomics. Nature Methods22(2), 287–297 (2025)
work page 2025
-
[14]
Devlin, J., Chang, M.W., Lee, K., Toutanova, K.: Bert: Pre-training of deep bidirectional transformers for language understanding. In: Proceedings of NAACL-HLT. Association for Computational Linguistics (2019)
work page 2019
-
[15]
In: Proceedings of the 58th Annual Meeting of the Association for Computational Linguistics
DeYoung, J., Jain, S., Rajani, N.F., Lehman, E., Xiong, C., Socher, R., Wallace, B.C.: ERASER: A benchmark to evaluate rationalized NLP models. In: Proceedings of the 58th Annual Meeting of the Association for Computational Linguistics. pp. 4443–4458 (2020)
work page 2020
-
[16]
Nature Reviews Genetics20(7), 389–403 (2019)
Eraslan, G., Avsec, Ž., Gagneur, J., Theis, F.J.: Deep learning: new computational modelling techniques for genomics. Nature Reviews Genetics20(7), 389–403 (2019)
work page 2019
-
[17]
BMC Genomic Data24(1), 25 (2023)
Grešová, K., Martinek, V ., ˇCechák, D., Šime ˇcek, P., Alexiou, P.: Genomic benchmarks: a collection of datasets for genomic sequence classification. BMC Genomic Data24(1), 25 (2023)
work page 2023
-
[18]
Genome biology8(2), R24 (2007)
Gupta, S., Stamatoyannopoulos, J.A., Bailey, T.L., Noble, W.S.: Quantifying similarity between motifs. Genome biology8(2), R24 (2007)
work page 2007
-
[19]
Journal of Machine Learning Research24(34), 1–11 (2023)
Hedström, A., Weber, L., Krakowczyk, D., Bareeva, D., Motzkus, F., Samek, W., Lapuschkin, S., Höhne, M.M.C.: Quantus: An explainable ai toolkit for responsible evaluation of neural network explanations and beyond. Journal of Machine Learning Research24(34), 1–11 (2023)
work page 2023
-
[20]
Bioinformatics37(15), 2112–2120 (2021)
Ji, Y ., Zhou, Z., Liu, H., Davuluri, R.V .: DNABERT: pre-trained Bidirectional Encoder Rep- resentations from Transformers model for DNA-language in genome. Bioinformatics37(15), 2112–2120 (2021)
work page 2021
-
[21]
In: Advances in Neural Information Processing Systems
Lundberg, S.M., Lee, S.I.: A Unified Approach to Interpreting Model Predictions. In: Advances in Neural Information Processing Systems. vol. 30. Curran Associates, Inc. (2017)
work page 2017
-
[22]
In: The Twelfth International Conference on Learning Representations (2024) 10
Marin, F.I., Teufel, F., Horlacher, M., Madsen, D., Pultz, D., Winther, O., Boomsma, W.: BEND: Benchmarking DNA Language Models on Biologically Meaningful Tasks. In: The Twelfth International Conference on Learning Representations (2024) 10
work page 2024
-
[23]
Explainable AI: interpreting, explaining and visualizing deep learning pp
Montavon, G., Binder, A., Lapuschkin, S., Samek, W., Müller, K.R.: Layer-Wise Relevance Propagation: An Overview. Explainable AI: interpreting, explaining and visualizing deep learning pp. 193–209 (2019)
work page 2019
-
[24]
Nature Reviews Genetics24(2), 125–137 (2023)
Novakovsky, G., Dexter, N., Libbrecht, M.W., Wasserman, W.W., Mostafavi, S.: Obtaining genetics insights from deep learning via explainable artificial intelligence. Nature Reviews Genetics24(2), 125–137 (2023)
work page 2023
-
[25]
Proceedings of the IEEE 109(3), 247–278 (2021)
Samek, W., Montavon, G., Lapuschkin, S., Anders, C.J., Müller, K.R.: Explaining Deep Neural Networks and Beyond: A Review of Methods and Applications. Proceedings of the IEEE 109(3), 247–278 (2021)
work page 2021
-
[26]
Nucleic acids research 32(suppl_1), D91–D94 (2004)
Sandelin, A., Alkema, W., Engström, P., Wasserman, W.W., Lenhard, B.: JASPAR: an open- access database for eukaryotic transcription factor binding profiles. Nucleic acids research 32(suppl_1), D91–D94 (2004)
work page 2004
-
[27]
In: Proceedings of the 34th International Conference on Machine Learning
Shrikumar, A., Greenside, P., Kundaje, A.: Learning Important Features Through Propagating Activation Differences. In: Proceedings of the 34th International Conference on Machine Learning. pp. 3145–3153. PMLR (2017)
work page 2017
- [28]
-
[29]
Genome Biology26(1), 203 (2025)
Tang, Z., Somia, N., Yu, Y ., Koo, P.K.: Evaluating the representational power of pre-trained DNA language models for regulatory genomics. Genome Biology26(1), 203 (2025)
work page 2025
-
[30]
Wolf, T., Debut, L., Sanh, V ., Chaumond, J., Delangue, C., Moi, A., Cistac, P., Rault, T., Louf, R., Funtowicz, M., Davison, J., Shleifer, S., von Platen, P., Ma, C., Jernite, Y ., Plu, J., Xu, C., Le Scao, T., Gugger, S., Drame, M., Lhoest, Q., Rush, A.: Transformers: State-of-the-art natural language processing. In: Proceedings of the 2020 conference o...
work page 2020
-
[31]
arXiv preprint arXiv:2306.15006 , year=
Zhou, Z., Ji, Y ., Li, W., Dutta, P., Davuluri, R., Liu, H.: Dnabert-2: Efficient foundation model and benchmark for multi-species genome. arXiv preprint arXiv:2306.15006 (2023)
-
[32]
Nature genetics51(1), 12–18 (2019) 11
Zou, J., Huss, M., Abid, A., Mohammadi, P., Torkamani, A., Telenti, A.: A primer on deep learning in genomics. Nature genetics51(1), 12–18 (2019) 11
work page 2019
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.