pANO-F12: An atomic natural orbital-inspired route to more compact basis sets for F12 explicitly correlated methods
Pith reviewed 2026-05-22 01:51 UTC · model grok-4.3
The pith
Energy minimization under linear independence constraints yields compact pANO-F12 basis sets equivalent in quality to larger F12 sets.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
An energy minimization-based contraction process under linear independence constraints yields pseudo-ANO (pANO) basis sets that are functionally equivalent in quality to those derived from first-order reduced density matrices while accounting for the F12 geminal. When this recipe is applied to F12 methods the resulting pANO-F12 basis sets, unlike cc-pVnZ-F12, exhibit the familiar shell structure seen in cc-pVnZ and ANO basis sets and offer a route to more compact F12 basis sets more amenable to medium-sized systems, especially in conjunction with localized pair natural orbital approaches. The pANO approach is most beneficial for the smaller double- and triple-zeta basis sets, offering either
What carries the argument
The pANO contraction process: an energy minimization under linear independence constraints that generates pseudo-atomic natural orbital basis sets functionally equivalent to density-matrix ANOs while incorporating the F12 geminal.
If this is right
- pANO-F12 basis sets exhibit the shell structure typical of cc-pVnZ and ANO families unlike the oversized cc-pVnZ-F12 sets.
- The new sets are more compact and therefore more practical for medium-sized molecules.
- Performance gains are largest when pANO-F12 is used together with localized pair natural orbital methods.
- At double- and triple-zeta levels the sets deliver either better results at fixed cost or equivalent results at reduced cost.
Where Pith is reading between the lines
- The contraction recipe could be applied to other explicitly correlated methods where density matrices are difficult to obtain.
- Smaller F12 basis sets may make high-accuracy calculations feasible for molecules larger than those currently treated routinely.
- Further tuning of the linear-independence constraints might produce sets optimized for particular molecular properties or local correlation schemes.
Load-bearing premise
That minimizing energy under linear independence constraints produces basis sets of quality equivalent to those obtained from first-order reduced density matrices for F12 methods.
What would settle it
A set of thermochemical or benzene out-of-plane vibration calculations in which pANO-F12 fails to match or beat the accuracy of cc-pVnZ-F12 at the same or lower computational cost would falsify the functional equivalence and compactness claims.
Figures
read the original abstract
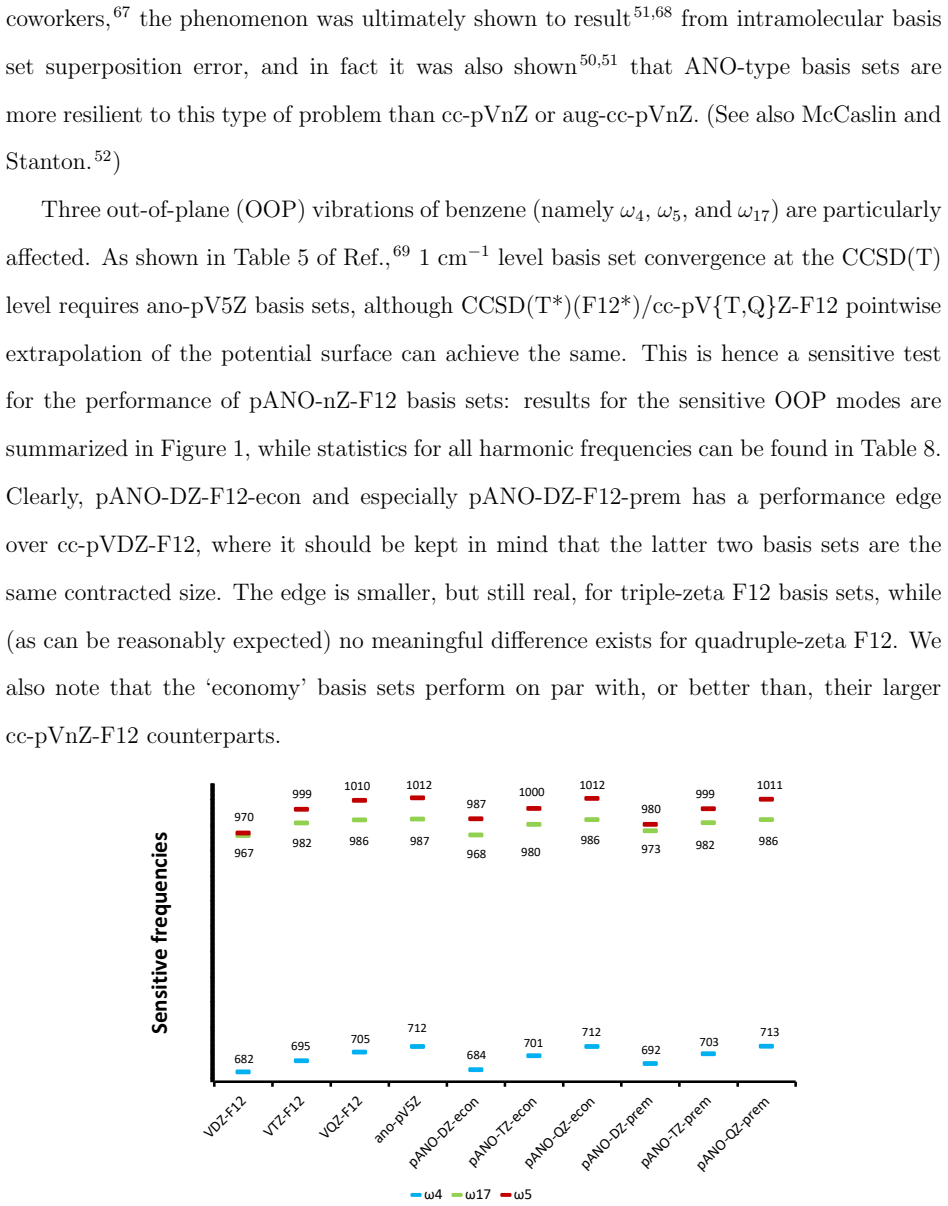
Explicitly correlated methods such as MP2-F12 and CCSD(F12*) exhibit much faster basis set convergence (asymptotically $\propto L^{-7}$, with L the highest angular momentum) than orbital-only approaches. Yet it has been pointed out that cc-pVnZ-F12 basis sets themselves are substantially larger than the corresponding cc-pVnZ, and specifically that cc-pVDZ-F12 is the size of cc-pVTZ. One way to generate compact basis sets in an orbital-only context are Atomic Natural Orbital (ANO) basis sets [J. Alml\"of and P. R. Taylor, JCP 86, 4070 (1987)]. However, obtaining the required first-order reduced density matrix while properly accounting for the F12 geminal is problematic. In this work, we show that an energy minimization-based contraction process under linear independence constraints yields `pseudo-ANO' (pANO) basis sets that are functionally equivalent in quality. Subsequently, we apply this recipe to obtain pANO-F12 basis sets from the same elements, then validate them for several thermochemical benchmarks and for the hypersensitive out-of-plane vibrations of benzene. We show that, unlike cc-pVnZ-F12, pANO-F12 exhibits the familiar shell structure seen in cc-pVnZ and ANO basis sets, and that pANO-F12 offers a route to more compact F12 basis sets more amenable to medium-sized systems, especially in conjunction with localized pair natural orbital approaches. Overall, the pANO approach is most beneficial for the smaller double-and triple-zeta basis sets, offering either superior performance to cc-pVnZ-F12 at same cost, or similar performance at lower cost.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes pANO-F12 basis sets constructed via an energy-minimization contraction procedure subject to linear-independence constraints. This approach is presented as a workaround for the difficulty of obtaining an F12-adjusted first-order reduced density matrix, yielding 'pseudo-ANO' sets that the authors claim are functionally equivalent in quality to conventional ANO-F12 constructions. The new basis sets are applied to thermochemical benchmarks and the out-of-plane vibrations of benzene; the authors report that, unlike cc-pVnZ-F12, the pANO-F12 families recover the familiar shell structure of cc-pVnZ and ANO sets and are especially advantageous for double- and triple-zeta regimes when used with localized pair-natural-orbital methods.
Significance. If the claimed functional equivalence is rigorously established, the work supplies a practical route to compact explicitly correlated basis sets that retain the rapid basis-set convergence of F12 methods while reducing the size penalty that currently makes cc-pVnZ-F12 sets larger than their orbital-only counterparts. This would be particularly useful for medium-sized molecules treated with local correlation techniques. The energy-minimization route also avoids the technical obstacle of constructing a geminal-aware density matrix, which is a genuine methodological contribution.
major comments (1)
- [Abstract / pANO construction] Abstract and the description of the pANO construction: the central claim that an energy-minimization procedure under linear-independence constraints produces pseudo-ANOs that are functionally equivalent to those derived from an F12-adjusted 1-RDM is load-bearing for the entire paper. The manuscript must demonstrate (or provide a clear argument) that the chosen energy functional at the MP2-F12 level embeds the same geminal-dependent correlation contributions that would appear in a proper F12 density matrix; otherwise the stationary orbital occupancies and contraction coefficients may systematically differ, especially in the double- and triple-zeta regimes where the largest practical benefit is asserted.
minor comments (2)
- [Abstract] The abstract refers to 'several thermochemical benchmarks' and 'hypersensitive out-of-plane vibrations of benzene' but supplies neither the specific data sets, error statistics, nor exclusion criteria. Adding a concise table or figure with these results (including error bars and direct comparison to cc-pVnZ-F12) would make the validation section more transparent.
- [Method] Notation for the linear-independence constraints and the precise form of the energy functional used in the contraction step should be defined explicitly (ideally with an equation) rather than left at the level of a verbal description.
Simulated Author's Rebuttal
We thank the referee for their careful reading of the manuscript and for the constructive major comment. We address the point directly below and have revised the manuscript to strengthen the justification for the pANO construction.
read point-by-point responses
-
Referee: [Abstract / pANO construction] Abstract and the description of the pANO construction: the central claim that an energy-minimization procedure under linear-independence constraints produces pseudo-ANOs that are functionally equivalent to those derived from an F12-adjusted 1-RDM is load-bearing for the entire paper. The manuscript must demonstrate (or provide a clear argument) that the chosen energy functional at the MP2-F12 level embeds the same geminal-dependent correlation contributions that would appear in a proper F12 density matrix; otherwise the stationary orbital occupancies and contraction coefficients may systematically differ, especially in the double- and triple-zeta regimes where the largest practical benefit is asserted.
Authors: We appreciate the referee highlighting this foundational aspect of the pANO approach. The energy-minimization procedure optimizes contraction coefficients by directly minimizing the MP2-F12 energy for atomic and small-molecular prototypes while enforcing linear independence. Because the MP2-F12 energy expression explicitly incorporates the geminal-dependent F12 correction terms (both the complementary auxiliary basis set and the explicit correlation contributions), the stationary points with respect to the orbital coefficients inherently weight orbitals according to their importance for these geminal contributions. This supplies a clear argument that the chosen functional embeds the relevant correlation physics without requiring an explicit F12-adjusted 1-RDM. While the resulting occupancies are not formally identical to those from a density-matrix diagonalization, the practical outcome is functional equivalence, as confirmed by the thermochemical and vibrational benchmarks. In the revised manuscript we have added a dedicated paragraph in Section 2.2 that spells out this reasoning, discusses why systematic deviations are not expected in the DZ/TZ regimes, and reports additional numerical checks on orbital character. revision: yes
Circularity Check
Energy-minimization contraction is an independent alternative to F12-adjusted 1-RDM
full rationale
The paper's central step replaces the problematic F12-adjusted first-order reduced density matrix with an energy-minimization procedure under linear-independence constraints. This construction is presented as a distinct recipe rather than a reparameterization or fit to target F12 results. No load-bearing premise reduces to a self-citation, no uniqueness theorem is imported from the authors' prior work, and no prediction is shown to be equivalent to its inputs by construction. Validation on thermochemical benchmarks and benzene vibrations is offered as external evidence of functional equivalence, keeping the derivation self-contained against the stated inputs.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Energy minimization under linear independence constraints produces basis sets functionally equivalent to density-matrix ANOs for F12 methods
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
an energy minimization-based contraction process under linear independence constraints yields 'pseudo-ANO' (pANO) basis sets that are functionally equivalent in quality
-
IndisputableMonolith/Foundation/DimensionForcing.leanalexander_duality_circle_linking unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
pANO-F12 exhibits the familiar shell structure seen in cc-pVnZ and ANO basis sets
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
Hylleraas, E. A. Neue Berechnung der Energie des Heliums im Grundzustande des tiefsten Terms von Ortho-Helium. Zeitschrift f \"u r Physik 1929, 54, 347--366
work page 1929
-
[2]
Pekeris, C. L. Ground State of Two-Electron Atoms. Physical Review 1958, 112, 1649--1658
work page 1958
-
[3]
Pekeris, C. L. 1^ 1 S and 2^ 3 S States of Helium. Physical Review 1959, 115, 1216--1221
work page 1959
-
[4]
Pekeris, C. L. 1^ 1 S , 2^ 1 S , and 2^ 3 S States of H ^ - and of He. Physical Review 1962, 126, 1470--1476
work page 1962
-
[5]
Pekeris, C. L. Excited S States of Helium. Physical Review 1962, 127, 509--518
work page 1962
-
[6]
Boys , S. F. Electronic Wave Functions. I. A General Method of Calculation for the Stationary States of Any Molecular System . Proceedings of the Royal Society of London Series A 1950, 200, 542--554
work page 1950
-
[7]
Boys, S. F.; Handy, N. C. The determination of energies and wavefunctions with full electronic correlation. Proceedings of the Royal Society of London. Series A. Mathematical and Physical Sciences 1969, 310, 43--61
work page 1969
-
[8]
Boys, S. F.; Handy, N. C. A condition to remove the indeterminacy in interelectronic correlation functions. Proceedings of the Royal Society of London. Series A. Mathematical and Physical Sciences 1969, 309, 209--220
work page 1969
-
[9]
Boys, S. F.; Handy, N. C. A calculation for the energies and wavefunctions for states of neon with full electronic correlation accuracy. Proceedings of the Royal Society of London. Series A. Mathematical and Physical Sciences 1969, 310, 63--78
work page 1969
-
[10]
Boys, S. F.; Handy, N. C. A first solution, for LiH, of a molecular transcorrelated wave equation by means of restricted numerical integration. Proceedings of the Royal Society of London. Series A. Mathematical and Physical Sciences 1969, 311, 309--329
work page 1969
-
[11]
Many-Body Problem with Strong Forces
Jastrow, R. Many-Body Problem with Strong Forces. Phys. Rev. 1955, 98, 1479--1484
work page 1955
-
[12]
Handy, N. C. The transcorrelated method for accurate correlation energies using gaussian-type functions: examples on He, H _2 , LiH and H _2 O. Molecular Physics 1972, 23, 1--27
work page 1972
-
[13]
Schraivogel, T.; Cohen, A. J.; Alavi, A.; Kats, D. Transcorrelated coupled cluster methods. J. Chem. Phys. 2021, 155, 191101
work page 2021
-
[14]
Kutzelnigg, W.; Klopper, W. Wave functions with terms linear in the interelectronic coordinates to take care of the correlation cusp. I . General theory. J. Chem. Phys. 1991, 94, 1985--2001
work page 1991
-
[15]
Termath, V.; Klopper, W.; Kutzelnigg, W. Wave functions with terms linear in the interelectronic coordinates to take care of the correlation cusp. II . M ller--Plesset calculations for the singlet states of He and H _2 . J. Chem. Phys. 1991, 94, 2002--2019
work page 1991
-
[16]
Noga, J.; Kutzelnigg, W. Coupled-cluster theory that takes care of the correlation cusp by inclusion of linear terms in the interelectronic coordinates. J. Chem. Phys. 1994, 101, 7738--7755
work page 1994
-
[17]
Rates of convergence of the partial-wave expansions of atomic correlation energies
Kutzelnigg, W.; Morgan, I., John D. Rates of convergence of the partial-wave expansions of atomic correlation energies. J. Chem. Phys. 1992, 96, 4484--4508
work page 1992
-
[18]
r_ 12 -dependent terms in the wave function as closed sums of partial wave amplitudes for large l
Kutzelnigg, W. r_ 12 -dependent terms in the wave function as closed sums of partial wave amplitudes for large l . Theor. Chem. Acc. 1985, 68, 445--469
work page 1985
-
[19]
Natural Spin Orbital Analysis of Hydrogen Molecule Wave Functions
Shull, H. Natural Spin Orbital Analysis of Hydrogen Molecule Wave Functions. J. Chem. Phys. 1959, 30, 1405--1413
work page 1959
-
[20]
Klopper, W.; Samson, C. C. M. Explicitly correlated second-order M ller--Plesset methods with auxiliary basis sets. J. Chem. Phys. 2002, 116, 6397--6410
work page 2002
-
[21]
Valeev, E. F. Improving on the resolution of the identity in linear R12 ab initio theories. Chem. Phys. Lett. 2004, 395, 190--195
work page 2004
-
[22]
Initiation of explicitly correlated Slater-type geminal theory
Ten-no, S. Initiation of explicitly correlated Slater-type geminal theory. Chem. Phys. Lett. 2004, 398, 56--61
work page 2004
-
[23]
Werner, H.-J.; Knowles, P. J.; Manby, F. R.; Black, J. A.; Doll, K.; He elmann, A.; Kats, D.; K \"o hn, A.; Korona, T.; Kreplin, D. A. et al. The Molpro quantum chemistry package. J. Chem. Phys. 2020, 152, 144107
work page 2020
-
[24]
Balasubramani, S. G.; Chen, G. P.; Coriani, S.; Diedenhofen, M.; Frank, M. S.; Franzke, Y. J.; Furche, F.; Grotjahn, R.; Harding, M. E.; H \"a ttig, C. et al. TURBOMOLE : Modular program suite for ab initio quantum-chemical and condensed-matter simulations. J. Chem. Phys. 2020, 152, 184107
work page 2020
-
[25]
Franzke, Y. J.; Holzer, C. H.; Andersen, J. H.; Begu s i \'c , T.; Bruder, F.; Coriani, S.; Della Sala, F.; Fabiano, E.; Fedotov, D. A.; F \"u rst, S. et al. TURBOMOLE : Today and Tomorrow. J. Chem. Theory Comput. 2023, 19, 6859--6890
work page 2023
-
[26]
R.; Mester, D.; Rolik, Z.; Samu, G.; Csontos, J.; Cs \'o ka, J.; Szab \'o , P
K \'a llay, M.; Nagy, P. R.; Mester, D.; Rolik, Z.; Samu, G.; Csontos, J.; Cs \'o ka, J.; Szab \'o , P. B.; Gyevi-Nagy, L.; H \'e gely, B. et al. The MRCC program system: Accurate quantum chemistry from water to proteins. J. Chem. Phys. 2020, 152, 074107
work page 2020
-
[27]
R.; Cs \'o ka, J.; Gyevi-Nagy, L.; Szab \'o , P
Mester, D.; Nagy, P. R.; Cs \'o ka, J.; Gyevi-Nagy, L.; Szab \'o , P. B.; Horv \'a th, R. A.; Petrov, K.; H \'e gely, B.; Lad \'o czki, B.; Samu, G. et al. Overview of Developments in the MRCC Program System. J. Phys. Chem. A 2025, 129, 2086--2107
work page 2025
-
[28]
Persson, B. J.; Taylor, P. R. Accurate quantum-chemical calculations: The use of Gaussian-type geminal functions in the treatment of electron correlation. J. Chem. Phys. 1996, 105, 5915--5926
work page 1996
-
[29]
H \"a ttig, C.; Klopper, W.; K \"o hn, A.; Tew, D. P. Explicitly Correlated Electrons in Molecules. Chem. Rev. 2012, 112, 4--74
work page 2012
-
[30]
Kong, L.; Bischoff, F. A.; Valeev, E. F. Explicitly Correlated R12 / F12 Methods for Electronic Structure. Chem. Rev. 2012, 112, 75--107
work page 2012
-
[31]
Valeev, E. F.; Sherrill, C. D. Perspective: Explicitly correlated electronic structure theory for molecules. J. Chem. Phys. 2017, 146, 080901
work page 2017
-
[32]
Explicitly correlated local coupled-cluster methods using pair natural orbitals
Ma, Q.; Werner, H.-J. Explicitly correlated local coupled-cluster methods using pair natural orbitals. Wiley Interdisciplinary Reviews: Computational Molecular Science 2018, 8, e1371
work page 2018
-
[33]
Tew, D. P. Principal domains in F12 explicitly correlated theory. Advances in Quantum Chemistry 2021, 83, 83--106
work page 2021
-
[34]
Mehta, N.; Martin, J. M. L. Explicitly Correlated Double-Hybrid DFT: A Comprehensive Analysis of the Basis Set Convergence on the GMTKN55 Database. J. Chem. Theory Comput. 2022, 18, 5978--5991
work page 2022
-
[35]
Mehta, N.; Martin, J. M. L. Reduced-Scaling Double Hybrid Density Functional Theory with Rapid Basis Set Convergence through Localized Pair Natural Orbital F12. J. Phys. Chem. Lett. 2022, 13, 9332--9338
work page 2022
-
[36]
Martin, J. M. L.; Santra, G. Empirical Double‐Hybrid Density Functional Theory: A ‘Third Way’ in Between WFT and DFT. Israel Journal of Chemistry 2020, 60, 787--804, [Special Issue:Computational Materials Science in Israel]
work page 2020
-
[37]
Peterson, K. A.; Adler, T. B.; Werner, H.-J. Systematically convergent basis sets for explicitly correlated wavefunctions: the atoms H, He, B--Ne, and Al--Ar. J. Chem. Phys. 2008, 128, 084102
work page 2008
-
[38]
Dunning Jr, T. H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007--1023
work page 1989
-
[39]
Peterson, K. A. In Encyclopedia of Inorganic and Bioinorganic Chemistry; Scott, R. A., Ed.; John Wiley & Sons, Ltd., 2011
work page 2011
-
[40]
Jensen, F. Atomic orbital basis sets. Wiley Interdisciplinary Reviews: Computational Molecular Science 2013, 3, 273--295
work page 2013
-
[41]
Hill, J. G. Gaussian Basis Sets for Molecular Applications. International Journal of Quantum Chemistry 2013, 113, 21--34
work page 2013
-
[42]
In Reviews in Computational Chemistry; Parrill, A
Nagy, B.; Jensen, F. In Reviews in Computational Chemistry; Parrill, A. L., Lipkowitz, K. B., Eds.; Wiley, 2017; Vol. 30; pp 93--150
work page 2017
-
[43]
Sylvetsky, N.; Peterson, K. A.; Karton, A.; Martin, J. M. L. Toward a W4-F12 approach: Can explicitly correlated and orbital-based ab initio CCSD(T) limits be reconciled? J. Chem. Phys. 2016, 144, 214101
work page 2016
-
[44]
Relativistic Pseudopotentials : Their Development and Scope of Applications
Dolg, M.; Cao, X. Relativistic Pseudopotentials : Their Development and Scope of Applications. Chem. Rev. 2012, 112, 403--480
work page 2012
-
[45]
Hill, J. G.; Peterson, K. A. Correlation consistent basis sets for explicitly correlated post-d main group elements. J. Chem. Phys. 2014, 141, 094106
work page 2014
-
[46]
Hill, J. G.; Shaw, R. A. Pseudopotential-based basis sets for the group 11 (Cu, Ag, Au) and 12 (Zn, Cd, Hg) elements. J. Chem. Phys. 2021, 155, 174113
work page 2021
-
[47]
Semidalas, E.; Martin, J. M. L. Correlation Consistent Basis Sets for Explicitly Correlated Theory: The Transition Metals. J. Chem. Theory Comput. 2023, 19, 5806–5820
work page 2023
-
[48]
Peterson, K. A.; Kesharwani, M. K.; Martin, J. M. L. The cc-pV5Z-F12 basis set: reaching the basis set limit in explicitly correlated calculations. Mol. Phys. 2015, 113, 1551--1558
work page 2015
-
[49]
Alml \"o f, J.; Taylor, P. R. General contraction of Gaussian basis sets. I. Atomic natural orbitals for first- and second-row atoms. J. Chem. Phys. 1987, 86, 4070--4077
work page 1987
-
[50]
Martin, J. M. L.; Taylor, P. R.; Lee, T. J. The harmonic frequencies of benzene. A case for atomic natural orbital basis sets. Chem. Phys. Lett. 1997, 275, 414--422
work page 1997
-
[51]
Martin, J. M. L.; Lee, T. J.; Taylor, P. R. A purely ab initio spectroscopic quality quartic force field for acetylene. J. Chem. Phys. 1998, 108, 676--691
work page 1998
-
[52]
McCaslin, L.; Stanton, J. F. Calculation of fundamental frequencies for small polyatomic molecules: a comparison between correlation consistent and atomic natural orbital basis sets. Mol. Phys. 2013, 111, 1492--1496
work page 2013
-
[53]
Neese, F.; Valeev, E. F. Revisiting the atomic natural orbital approach for basis sets. J. Chem. Theory Comput. 2011, 7, 33--43
work page 2011
-
[54]
K \"o hn, A.; Tew, D. P. Accurate and efficient approximations to explicitly correlated coupled-cluster singles and doubles theory. J. Chem. Phys. 2010, 132, 231102
work page 2010
-
[55]
Coupled Cluster Wave Functions and a Dispersion-Weighted MP2 Method
Marchetti, O.; Werner, H.-J. Coupled Cluster Wave Functions and a Dispersion-Weighted MP2 Method . J. Phys. Chem. A 2009, 113, 11580--11585
work page 2009
-
[56]
Nelder, J. A.; Mead, R. A simplex method for function minimization. The Computer Journal 1965, 7, 308--313
work page 1965
-
[57]
Mehta, N.; Martin, J. M. L. MP2-F12 Basis Set Convergence near the Complete Basis Set Limit: Are h Functions Sufficient? J. Phys. Chem. A 2022, 126, 3964--3971
work page 2022
-
[58]
Weigend, F. A fully direct RI-HF algorithm: Implementation, optimised auxiliary basis sets, demonstration of accuracy and efficiency. Phys. Chem. Chem. Phys. 2002, 4, 4285--4291
work page 2002
-
[59]
Hill, J. G.; Peterson, K. A.; Knizia, G.; Werner, H.-J. Extrapolating MP2 and CCSD explicitly correlated correlation energies to the complete basis set limit with first and second row correlation consistent basis sets. J. Chem. Phys. 2009, 131, 194105
work page 2009
-
[60]
Nash, H. W.; Shaw, R. A.; Hill, J. G. Correlation consistent auxiliary basis sets in density fitting Hartree--Fock: The atoms sodium through argon revisited. J. Comput. Chem. 2023, 44, 1153--1167
work page 2023
-
[61]
Ranasinghe, D. S.; Petersson, G. A. CCSD(T)/CBS atomic and molecular benchmarks for H through Ar. J. Chem. Phys. 2013, 138, 144104
work page 2013
-
[62]
Karton, A.; Sylvetsky, N.; Martin, J. M. L. W4-17: A diverse and high-confidence dataset of atomization energies for benchmarking high-level electronic structure methods. J. Comput. Chem. 2017, 38, 2063--2075
work page 2017
-
[63]
Rez \'a c, J.; Riley, K. E.; Hobza, P. S66: A well-balanced database of benchmark interaction energies relevant to biomolecular structures. J. Chem. Theory Comput. 2011, 7, 2427--2438
work page 2011
-
[64]
Persson, B. J.; Taylor, P. R.; Lee, T. J. Ab initio geometry, quartic force field, and vibrational frequencies for P _4 . J. Chem. Phys. 1997, 107, 5051--5057
work page 1997
-
[65]
Barman, A.; Jones, G. H.; Weflen, K. E.; Shepelenko, M.; Martin, J. M. L. Coupling between thermochemical contributions of subvalence correlation and of higher-order post-CCSD(T) correlation effects --- a step toward `W5 theory'. J. Phys. Chem. A 2026, 130, 2943--2955
work page 2026
-
[66]
Simandiras, E. D.; Rice, J. E.; Lee, T. J.; Amos, R. D.; Handy, N. C. On the necessity of f basis functions for bending frequencies. J. Chem. Phys. 1988, 88, 3187--3195
work page 1988
-
[67]
Moran, D.; Simmonett, A. C.; Leininger, M. L.; Allen, W. D.; Schaefer, H. F.; von Ragu\'e Schleyer, P. Popular Theoretical Methods Predict Benzene and Arenes To Be Nonplanar. J. Am. Chem. Soc. 2006, 128, 9342--9343
work page 2006
-
[68]
Semidalas, E.; Martin, J. M. L. Can G4-like Composite Ab Initio Methods Accurately Predict Vibrational Harmonic Frequencies? Mol. Phys. 2024, 122, e2263593, [Timothy J. Lee memorial issue]
work page 2024
-
[69]
Kesharwani, M. K.; Sylvetsky, N.; Martin, J. M. L. The aug-cc-pVnZ-F12 basis set family: Correlation consistent basis sets for explicitly correlated benchmark calculations on anions and noncovalent complexes. J. Chem. Phys. 2017, 147
work page 2017
-
[70]
Widmark, P. O.; Malmqvist, P. .; Roos, B. O. Density matrix averaged atomic natural orbital (ANO) basis sets for correlated molecular wave functions - I. First row atoms . Theor. Chem. Acc. 1990, 77, 291--306
work page 1990
-
[71]
Alml \"o f, J.; Taylor, P. R. General contraction of Gaussian basis sets. II. Atomic natural orbitals and the calculation of atomic and molecular properties. J. Chem. Phys. 1990, 92, 551--560
work page 1990
-
[72]
Kendall, R. A.; Dunning, T. H.; Harrison, R. J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions . J. Chem. Phys. 1992, 96, 6796--6806
work page 1992
-
[73]
K.; Sylvetsky, N.; Köhn, A.; Tew, D
Kesharwani, M. K.; Sylvetsky, N.; Köhn, A.; Tew, D. P.; Martin, J. M. L. Do CCSD and approximate CCSD-F12 variants converge to the same basis set limits? The case of atomization energies. J. Chem. Phys. 2018, 149, 154109
work page 2018
-
[74]
Manna, D.; Kesharwani, M. K.; Sylvetsky, N.; Martin, J. M. L. Conventional and Explicitly Correlated ab Initio Benchmark Study on Water Clusters: Revision of the BEGDB and WATER27 Data Sets. J. Chem. Theory Comput. 2017, 13, 3136--3152 mcitethebibliography
work page 2017
- [75]
-
[76]
Electronic Wave Functions. I. A General Method of Calculation for the Stationary States of Any Molecular System. Proceedings of the Royal Society of London Series A , year = 1950, month = feb, volume =
work page 1950
-
[77]
Pekeris, C. L. , title =. Physical Review , year =
-
[78]
Many-Body Problem with Strong Forces , author =. Phys. Rev. , volume =. 1955 , month =. doi:10.1103/PhysRev.98.1479 , url =
-
[79]
Boys, S. F. and Handy, N. C. , title =. Proceedings of the Royal Society of London. Series A. Mathematical and Physical Sciences , year =
-
[80]
Handy, N. C. , title =. 1969 , volume =
work page 1969
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.