How Atoms Interact Within Molecules
Pith reviewed 2026-06-29 09:13 UTC · model grok-4.3
The pith
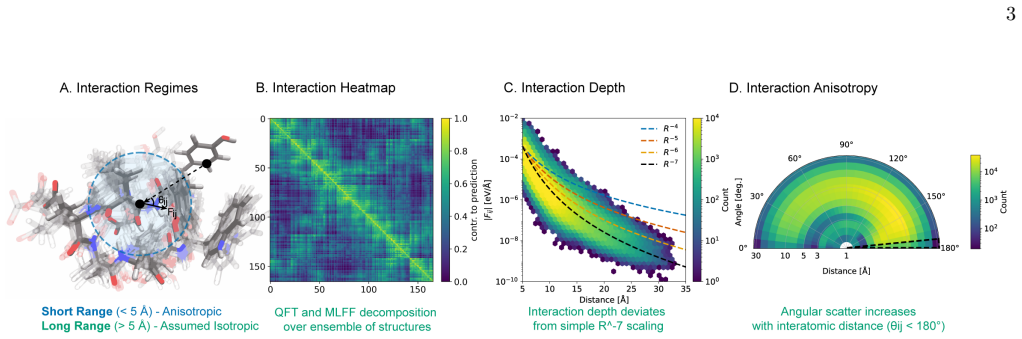
Interatomic forces in large molecules keep substantial scatter and anisotropy that grows with size.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
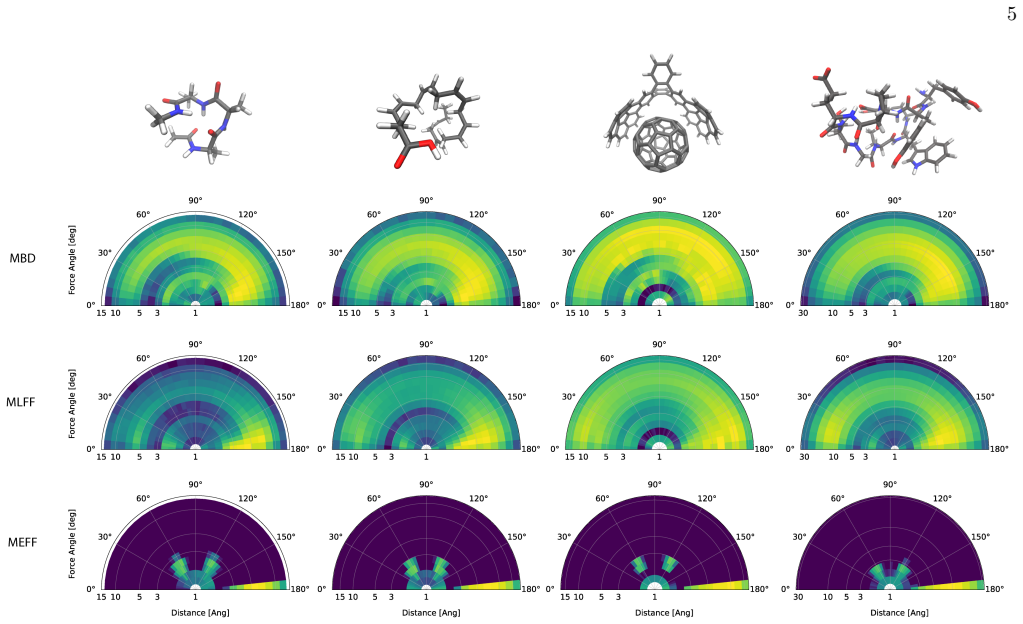
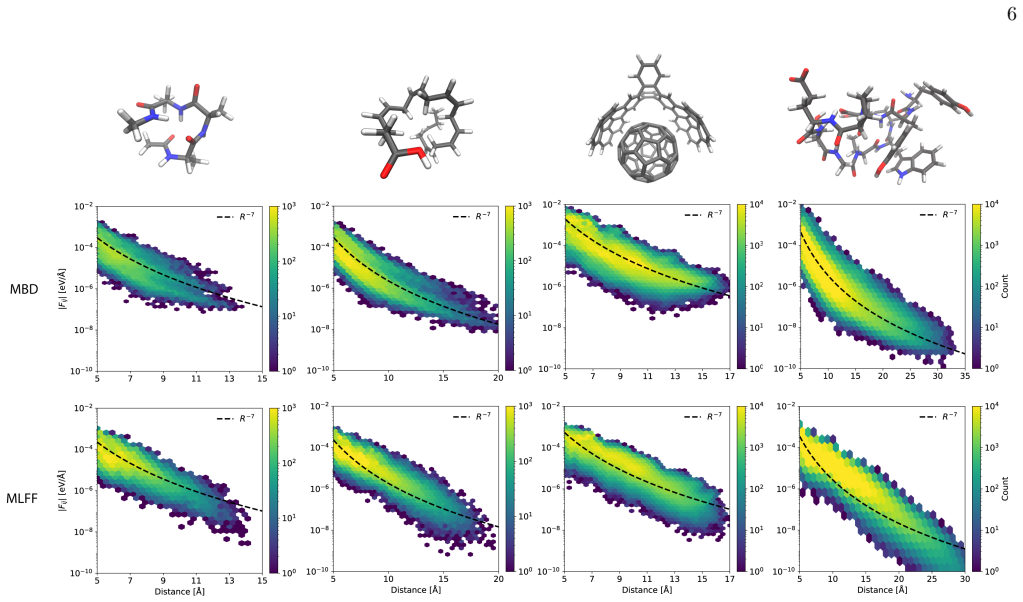
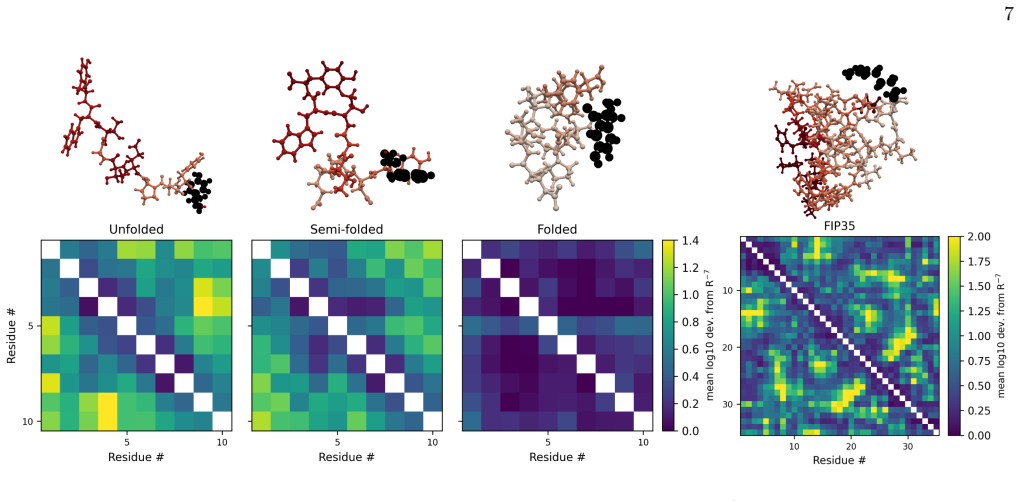
While the average interaction strength decays polynomially with interatomic separation, the interaction scatter remains robust and exhibits substantial anisotropy; both QFT and MLFF calculations show that increasing the molecular size further amplifies this scatter and anisotropy, a feature absent from traditional textbook empirical models.
What carries the argument
Direct computation of force depth and scatter via quantum field theory for long-range electron correlation combined with machine learning force fields applied to systems of hundreds of atoms.
If this is right
- Force models must incorporate size-amplified scatter to reach chemical accuracy in large systems.
- Attention should move from uniform atom-pair interactions to localized interacting hotspots that may control polymer folding pathways.
- The observed behavior explains why machine learning force fields succeed where simpler empirical forms fail.
- Development of future force fields should prioritize quantum-aware treatments of many-body anisotropy.
Where Pith is reading between the lines
- Hotspot-driven folding could alter predicted structures and dynamics of proteins and other biopolymers.
- The same size-dependent anisotropy may appear in condensed-phase or material systems beyond isolated molecules.
- Systematic checks on molecules of increasing size with independent electronic-structure methods would test the amplification trend.
Load-bearing premise
Quantum field theory and the machine learning force fields used here correctly compute the depth and scatter of interatomic forces in molecules with hundreds of atoms.
What would settle it
An independent high-accuracy calculation or measurement on a molecule of several hundred atoms that finds no growth in force scatter or anisotropy with increasing size would falsify the central claim.
Figures
read the original abstract
Fundamental understanding of interatomic forces in molecules must emerge from quantum mechanics, yet widely used empirical force fields rely on simplified mechanistic approximations that often fail to capture the complexity of many-body systems. Here we employ recent developments in quantum field theory (QFT) for long-range electron correlation and machine learning force fields (MLFFs) to directly compute the depth and scatter of interatomic forces for molecular systems containing hundreds of atoms. We find that while the average interaction strength decays polynomially with interatomic separation, the interaction scatter remains robust and exhibits substantial anisotropy. Both QFT and MLFFs demonstrate that increasing the molecular size further amplifies this scatter and anisotropy -- a phenomenon not considered in traditional textbook empirical models. These results provide new benchmarks for force models, shift the focus from interacting atoms to interacting ``hotspots'' that might determine the folding pathways of (bio)polymers, and rationalize why MLFFs are uniquely successful in capturing the nuances of complex molecular systems. Our findings offer a roadmap for the development of more accurate and quantum-aware molecular force fields.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript claims that QFT treatments of long-range electron correlation and MLFFs applied to molecules containing hundreds of atoms show that average interatomic interaction strength decays polynomially with separation while the scatter and anisotropy of those interactions remain robust and increase with system size, a feature absent from traditional empirical force fields; the results are said to supply new benchmarks, redirect attention to interaction hotspots, and rationalize MLFF performance.
Significance. If the reported size-dependent amplification of scatter is shown to be free of methodological artifacts, the work would supply a concrete, falsifiable critique of textbook force-field assumptions and a mechanistic rationale for why MLFFs outperform empirical models on complex systems. The absence of any reported validation against higher-level references, however, prevents the significance from being assessed at present.
major comments (2)
- [Abstract] Abstract: the claim that 'both QFT and MLFFs demonstrate that increasing the molecular size further amplifies this scatter and anisotropy' rests on the unverified assumption that the chosen QFT and MLFF approximations faithfully reproduce the depth, scatter, and size dependence of interatomic forces; no comparison to CCSD(T) on subsystems, no basis-set or cutoff convergence data, and no training-set-size tests are supplied to exclude the possibility that the reported anisotropy is an artifact.
- [Methods (implied)] No section supplies the explicit definition or numerical implementation of the 'scatter' metric whose size-dependent growth is the central observable; without this definition it is impossible to determine whether the reported amplification is independent of the computational protocol.
minor comments (1)
- [Abstract] The abstract should state the range of molecular sizes examined and name the specific QFT formulation and MLFF architectures employed.
Simulated Author's Rebuttal
We thank the referee for the careful and constructive review. The comments identify important points regarding validation and methodological clarity that we will address in revision. We respond to each major comment below.
read point-by-point responses
-
Referee: [Abstract] Abstract: the claim that 'both QFT and MLFFs demonstrate that increasing the molecular size further amplifies this scatter and anisotropy' rests on the unverified assumption that the chosen QFT and MLFF approximations faithfully reproduce the depth, scatter, and size dependence of interatomic forces; no comparison to CCSD(T) on subsystems, no basis-set or cutoff convergence data, and no training-set-size tests are supplied to exclude the possibility that the reported anisotropy is an artifact.
Authors: We acknowledge that the manuscript as submitted does not include direct CCSD(T) comparisons on subsystems or explicit convergence and training-set tests. While full CCSD(T) on the largest systems is computationally prohibitive, the QFT approach for long-range correlation and the MLFF architectures have been benchmarked against higher-level references in prior work on smaller molecules. To strengthen the manuscript and address the concern about possible artifacts, we will add a dedicated validation subsection reporting subsystem comparisons where feasible, basis-set and cutoff convergence data, and training-set-size sensitivity tests for the MLFFs. revision: yes
-
Referee: [Methods (implied)] No section supplies the explicit definition or numerical implementation of the 'scatter' metric whose size-dependent growth is the central observable; without this definition it is impossible to determine whether the reported amplification is independent of the computational protocol.
Authors: We agree that an explicit definition of the scatter metric is required for reproducibility and to confirm that the size dependence is not protocol-dependent. In the revised manuscript we will insert a new subsection in Methods that provides the precise mathematical definition of the scatter metric, its numerical implementation from the force data, and any relevant parameters used in its calculation. revision: yes
Circularity Check
No significant circularity; claims rest on independent QFT/MLFF computations
full rationale
The paper's derivation chain relies on applying external QFT developments for long-range electron correlation and MLFFs to compute interatomic force depth, scatter, and anisotropy in large molecules. The reported polynomial decay of average strength alongside robust scatter/anisotropy (amplified by system size) is presented as an output of these computations, not a definitional equivalence or fitted parameter renamed as prediction. No load-bearing self-citation chain, ansatz smuggling, or uniqueness theorem imported from prior author work is evident that would reduce the central observation to the inputs by construction. The abstract and described methodology treat QFT and MLFFs as independent tools providing new benchmarks, making the result falsifiable against higher-level references outside the present calculations. This is the expected non-finding for a paper whose core result is an empirical observation from external methods.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Frenkel and B
D. Frenkel and B. Smit,Understanding molecular simu- lation: from algorithms to applications(Elsevier, 2023)
2023
-
[2]
W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell, and P. A. Kollman, A second generation force field for the simulation of proteins, nucleic acids, and organic molecules, J. Am. Chem. Soc.117, 5179 (1995)
1995
-
[3]
A. D. MacKerell Jr, D. Bashford, M. Bellott, R. L. Dun- brack Jr, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha,et al., All-atom empirical potential for molecular modeling and dynamics studies of proteins, J. Phys. Chem. B.102, 3586 (1998)
1998
-
[4]
Stone,The theory of intermolecular forces(Oxford University Press, 2013)
A. Stone,The theory of intermolecular forces(Oxford University Press, 2013)
2013
-
[5]
Hermann, R
J. Hermann, R. A. DiStasio Jr, and A. Tkatchenko, First-principles models for van der waals interactions in molecules and materials: Concepts, theory, and applica- tions, Chem. Rev.117, 4714 (2017)
2017
-
[6]
Stone and A
A. Stone and A. Misquitta, Atom–atom potentials from ab initio calculations, Int. Rev. Phys. Chem.26, 193 (2007)
2007
-
[7]
Gurwitsch, ¨Uber die physiko-chemische attraktion- skraft, Z
L. Gurwitsch, ¨Uber die physiko-chemische attraktion- skraft, Z. physik. Chem.87, 323 (1914)
1914
-
[8]
Langmuir, The constitution and fundamental proper- ties of solids and liquids
I. Langmuir, The constitution and fundamental proper- ties of solids and liquids. ii. liquids., J. Am. Chem. Soc. 39, 1848 (1917)
1917
-
[9]
Stone and S
A. Stone and S. Price, Some new ideas in the theory of intermolecular forces: anisotropic atom-atom potentials, J. Phys. Chem.92, 3325 (1988)
1988
-
[10]
J. E. Jones, On the determination of molecular fields.—ii. from the equation of state of a gas, Proc. R. Soc. A.106, 463 (1924)
1924
-
[11]
London, The general theory of molecular forces, Trans
F. London, The general theory of molecular forces, Trans. Faraday Soc.33, 8b (1937)
1937
-
[12]
Tkatchenko and M
A. Tkatchenko and M. Scheffler, Accurate molecular van der waals interactions from ground-state electron den- sity and free-atom reference data, Phys. Rev. Lett.102, 073005 (2009)
2009
-
[13]
Grimme, J
S. Grimme, J. Antony, S. Ehrlich, and H. Krieg, A con- sistent and accurate ab initio parametrization of density functional dispersion correction (dft-d) for the 94 ele- ments h-pu, J. Chem. Phys.132, 154104 (2010)
2010
-
[14]
Dinur and A
U. Dinur and A. T. Hagler, Direct evaluation of non- bonding interactions from ab initio calculations, J. Am. Chem. Soc.111, 5149 (1989)
1989
-
[15]
Hauseux, T.-T
P. Hauseux, T.-T. Nguyen, A. Ambrosetti, K. S. Ruiz, S. P. Bordas, and A. Tkatchenko, From quantum to con- tinuum mechanics in the delamination of atomically-thin layers from substrates, Nat. Commun.11, 1651 (2020)
2020
-
[16]
Hauseux, A
P. Hauseux, A. Ambrosetti, S. P. Bordas, and A. Tkatchenko, Colossal enhancement of atomic force re- sponse in van der waals materials arising from many- body electronic correlations, Phys. Rev. Lett.128, 106101 (2022)
2022
-
[17]
M. J. Van Vleet, A. J. Misquitta, and J. Schmidt, New angles on standard force fields: Toward a general ap- proach for treating atomic-level anisotropy, J. Chem. Theory Comput.14, 739 (2018)
2018
-
[18]
Szalewicz, Symmetry-adapted perturbation theory of intermolecular forces, WIREs Comput
K. Szalewicz, Symmetry-adapted perturbation theory of intermolecular forces, WIREs Comput. Mol. Sci.2, 254 (2012)
2012
-
[19]
Kriz and D
K. Kriz and D. Van der Spoel, Quantification of anisotropy in exchange and dispersion interactions: A simple model for physics-based force fields, J. Phys. Chem. Lett.15, 9974 (2024)
2024
-
[20]
Schreiber and A
G. Schreiber and A. R. Fersht, Rapid, electrostatically assisted association of proteins, Nat. Struct. Mol. Biol. 3, 427 (1996)
1996
-
[21]
S. L. Price, Toward more accurate model intermolecular potentials for organic molecules, Rev. Comput. Chem. 14, 225 (2000)
2000
-
[22]
Honig and A
B. Honig and A. Nicholls, Classical electrostatics in biol- ogy and chemistry, Science268, 1144 (1995)
1995
-
[23]
P. Ren, J. Chun, D. G. Thomas, M. J. Schnieders, M. Marucho, J. Zhang, and N. A. Baker, Biomolecular electrostatics and solvation: a computational perspec- tive, Q. Rev. Biophys.45, 427 (2012)
2012
-
[24]
Zhou and X
H.-X. Zhou and X. Pang, Electrostatic interactions in protein structure, folding, binding, and condensation, Chem. Rev.118, 1691 (2018)
2018
-
[25]
Ambrosetti, N
A. Ambrosetti, N. Ferri, R. A. DiStasio Jr, and A. Tkatchenko, Wavelike charge density fluctuations and van der waals interactions at the nanoscale, Science351, 1171 (2016)
2016
-
[26]
St¨ ohr and A
M. St¨ ohr and A. Tkatchenko, Quantum mechanics of pro- teins in explicit water: The role of plasmon-like solute- solvent interactions, Sci. Adv.5, eaax0024 (2019)
2019
-
[27]
M. Gori, P. Kurian, and A. Tkatchenko, Second quanti- zation of many-body dispersion interactions for chemical and biological systems, Nat. Commun.14, 8218 (2023)
2023
-
[28]
R. Karimpour, M. Gori, and A. Tkatchenko, Quantum field approaches to chemical systems, arXiv preprint arXiv:2603.17582 (2026)
arXiv 2026
-
[29]
O. T. Unke, S. Chmiela, H. E. Sauceda, M. Gastegger, I. Poltavsky, K. T. Sch¨ utt, A. Tkatchenko, and K.-R. M¨ uller, Machine learning force fields, Chem. Rev.121, 10142 (2021)
2021
-
[30]
Tkatchenko, R
A. Tkatchenko, R. A. DiStasio Jr, R. Car, and M. Schef- fler, Accurate and efficient method for many-body van der waals interactions, Phys. Rev. Lett.108, 236402 (2012)
2012
-
[31]
R. A. DiStasio, V. V. Gobre, and A. Tkatchenko, Many- body van der waals interactions in molecules and con- densed matter, J. Phys.: Condens. Matter26, 213202 (2014)
2014
-
[32]
Sch¨ utt, O
K. Sch¨ utt, O. Unke, and M. Gastegger, Equivariant mes- sage passing for the prediction of tensorial properties and molecular spectra, Int. Conf. on Mach. Learn.139, 9377 (2021)
2021
-
[33]
Chmiela, V
S. Chmiela, V. Vassilev-Galindo, O. T. Unke, A. Kabylda, H. E. Sauceda, A. Tkatchenko, and K.-R. M¨ uller, Accurate global machine learning force fields for molecules with hundreds of atoms, Sci. Adv.9, eadf0873 (2023)
2023
-
[34]
T. Wang, X. He, M. Li, B. Shao, and T.-Y. Liu, Aimd- chig: Exploring the conformational space of a 166-atom protein chignolin with ab initio molecular dynamics, Sci. Data10, 549 (2023)
2023
-
[35]
Lindorff-Larsen, S
K. Lindorff-Larsen, S. Piana, R. O. Dror, and D. E. Shaw, How fast-folding proteins fold, Science334, 517 (2011)
2011
-
[36]
Honda, K
S. Honda, K. Yamasaki, Y. Sawada, and H. Morii, 10 residue folded peptide designed by segment statistics, 12 Structure12, 1507 (2004)
2004
-
[37]
Piana, K
S. Piana, K. Sarkar, K. Lindorff-Larsen, M. Guo, M. Gruebele, and D. E. Shaw, Computational design and experimental testing of the fastest-foldingβ-sheet pro- tein, J. Mol. Biol.405, 43 (2011)
2011
-
[38]
A. P. Jones, J. Crain, V. P. Sokhan, T. W. Whitfield, and G. J. Martyna, Quantum drude oscillator model of atoms and molecules: Many-body polarization and dispersion interactions for atomistic simulation, Phys. Rev. B87, 144103 (2013)
2013
-
[39]
Khabibrakhmanov, D
A. Khabibrakhmanov, D. V. Fedorov, A. Ambrosetti, J. Crain, K. L. Hunt, E. R. Johnson, K. D. Jordan, S. G´ oger, M. Gori, M. R. Karimpour,et al., Accu- rate noncovalent interactions in atomistic systems via quantum drude oscillators, J. Chem. Phys.163, 151001 (2025)
2025
-
[40]
Berezin,The method of second quantization(Aca- demic Press, 1966)
F. Berezin,The method of second quantization(Aca- demic Press, 1966)
1966
-
[41]
Blaizot and G
J.-P. Blaizot and G. Ripka,Quantum theory of finite sys- tems(MIT Press, 1986)
1986
-
[42]
R. F. W. Bader,Atoms in molecules: a quantum theory (Oxford University Press, 1990)
1990
-
[43]
Blanco, A
M. Blanco, A. Mart´ ın Pend´ as, and E. Francisco, Inter- acting quantum atoms: a correlated energy decomposi- tion scheme based on the quantum theory of atoms in molecules, J. Chem. Theory Comput.1, 1096 (2005)
2005
-
[44]
Bistoni, A
G. Bistoni, A. Altun, Z. Wang, and F. Neese, Local en- ergy decomposition analysis of london dispersion effects: From simple model dimers to complex biomolecular as- semblies, Acc. Chem. Res.57, 1411 (2024)
2024
-
[45]
K. T. Sch¨ utt, P. Kessel, M. Gastegger, K. A. Nicoli, A. Tkatchenko, and K.-R. M¨ uller, SchNetPack: A Deep Learning Toolbox For Atomistic Systems, J. Chem. The- ory Comput.15, 448 (2019)
2019
-
[46]
K. T. Sch¨ utt, S. S. P. Hessmann, N. W. A. Gebauer, J. Lederer, and M. Gastegger, SchNetPack 2.0: A neural network toolbox for atomistic machine learning, J. Chem. Phys.158, 144801 (2023)
2023
-
[47]
Esders, T
M. Esders, T. Schnake, J. Lederer, A. Kabylda, G. Mon- tavon, A. Tkatchenko, and K.-R. M¨ uller, Analyzing atomic interactions in molecules as learned by neural net- works, J. Chem. Theory Comput.21, 714 (2025)
2025
-
[48]
ˇSali, E
A. ˇSali, E. Shakhnovich, and M. Karplus, How does a protein fold?, Nature369, 248 (1994)
1994
-
[49]
H. N. Motlagh, J. O. Wrabl, J. Li, and V. J. Hilser, The ensemble nature of allostery, Nature508, 331 (2014)
2014
-
[50]
V. Blum, R. Gehrke, F. Hanke, P. Havu, V. Havu, X. Ren, K. Reuter, and M. Scheffler, Ab initio molecular simulations with numeric atom-centered orbitals, Com- put. Phys. Commun.180, 2175 (2009)
2009
-
[51]
X. Ren, P. Rinke, V. Blum, J. Wieferink, A. Tkatchenko, A. Sanfilippo, K. Reuter, and M. Scheffler, Resolution-of- identity approach to hartree–fock, hybrid density func- tionals, rpa, mp2 and gw with numeric atom-centered orbital basis functions, New J. Phys.14, 053020 (2012)
2012
-
[52]
Kabylda, J
A. Kabylda, J. T. Frank, S. Su´ arez-Dou, A. Khabibrakhmanov, L. Medrano Sandonas, O. T. Unke, S. Chmiela, K.-R. M¨ uller, and A. Tkatchenko, Molecular simulations with a pretrained neural network and universal pairwise force fields, J. Am. Chem. Soc. 147, 33723 (2025). 13 SUPPOR TING INFORMA TION TABLE S1. Dataset statistics and model errors for MBD refe...
2025
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.