The Language of Elution: Autoregressive Prediction of the Next Feature in Untargeted LC-HRMS Lipidomics
Pith reviewed 2026-06-28 07:45 UTC · model grok-4.3
The pith
Reversed-phase elution forms a predictable autoregressive sequence that models forecast from per-feature statistics alone at 98 percent top-1 accuracy.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
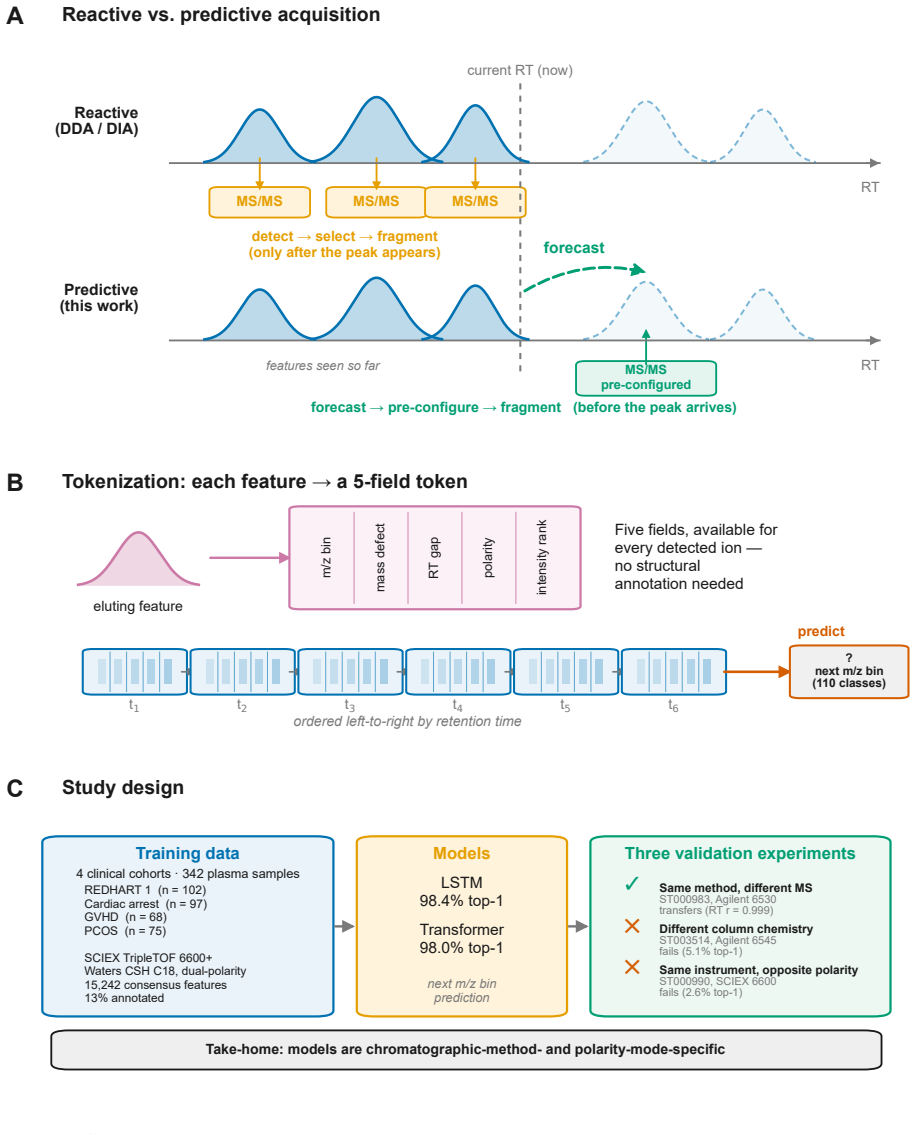
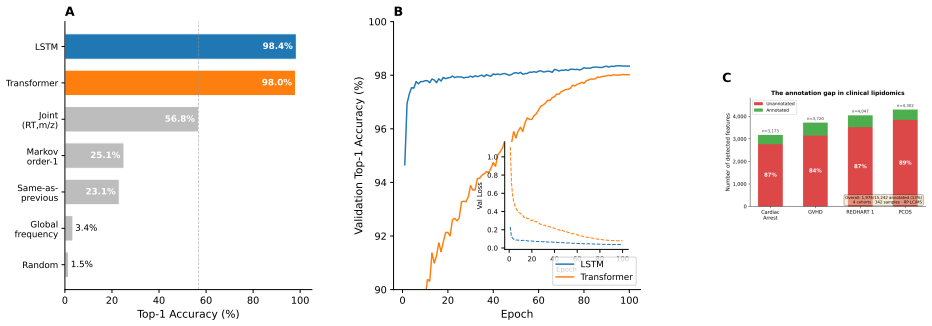
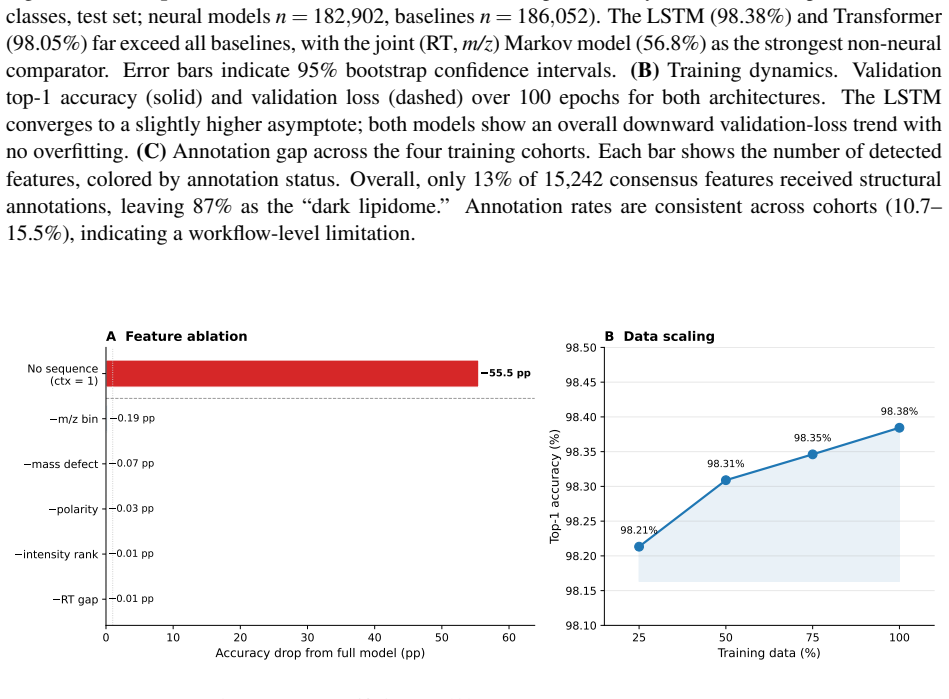
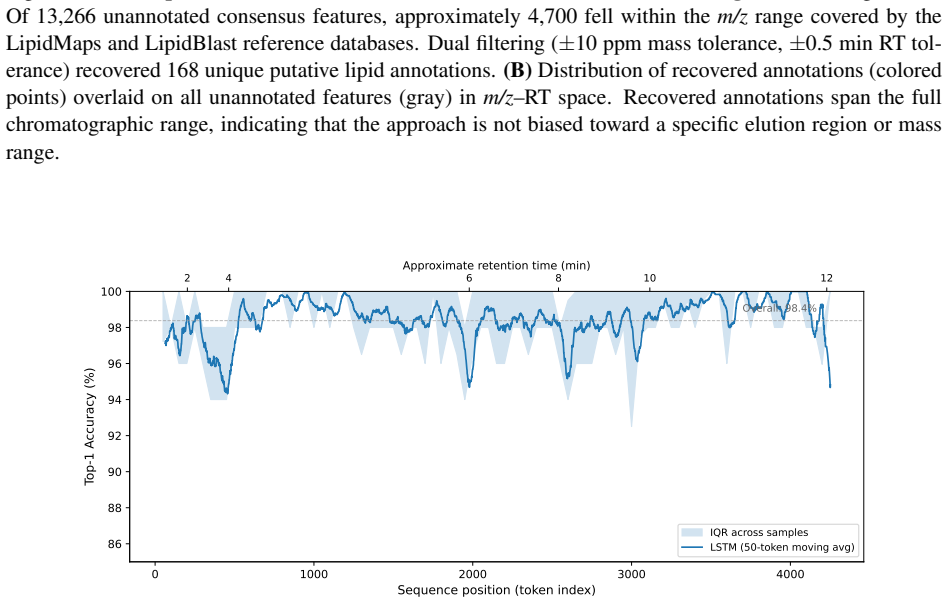
Treating elution as an autoregressive sequence task, LSTM and Transformer models trained on annotation-free per-feature statistics from four lipidomics cohorts predict the next m/z bin at 98.4 percent top-1 accuracy (99.99 percent top-5) and 98.0 percent respectively; ablation isolates autoregressive context as the dominant driver while no individual feature adds more than 0.2 percentage points, and transfer succeeds only under matched method and polarity.
What carries the argument
Autoregressive sequence models (LSTM and Transformer) that treat discretized m/z bins as tokens and predict the next bin from the five annotation-free per-token features.
If this is right
- Elution sequences are highly predictable from sequential context rather than individual molecular properties.
- Models trained on one instrument transfer with near-perfect correlation to another instrument using the identical chromatographic method.
- Accuracy collapses under altered column chemistry or polarity mode, confirming the prediction is method- and mode-specific.
- Fine-tuning on two to five quality-control injections restores nearly 50 percent top-1 accuracy after a condition change.
- The approach supplies a route to predictive rather than reactive MS/MS acquisition that could raise annotation coverage in untargeted lipidomics.
Where Pith is reading between the lines
- If elution order is this predictable, an instrument could pre-select the next precursor for fragmentation before the ion appears, directly addressing the reactive acquisition bottleneck.
- Each distinct LC method appears to possess its own characteristic elution grammar that must be learned separately.
- Because training uses only per-feature statistics, the same modeling strategy could be applied to other untargeted metabolomics datasets without requiring spectral libraries or structural annotations.
Load-bearing premise
That reversed-phase elution order forms a physically constrained autoregressive sequence governed primarily by hydrophobicity that can be learned from annotation-free per-feature statistics without reference to chemical identity.
What would settle it
Top-1 accuracy falling below 10 percent on a held-out set acquired with the same instrument but a different reversed-phase column chemistry would falsify the claim that the sequence is learnable and method-specific in the stated way.
Figures
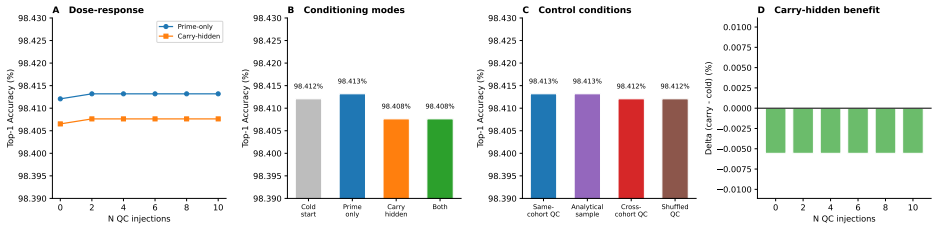
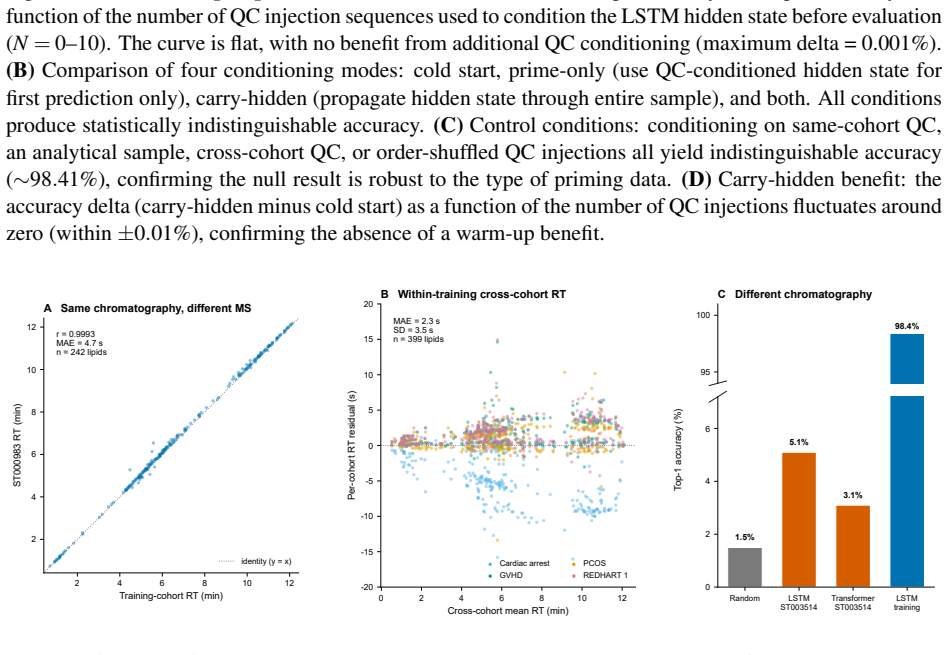
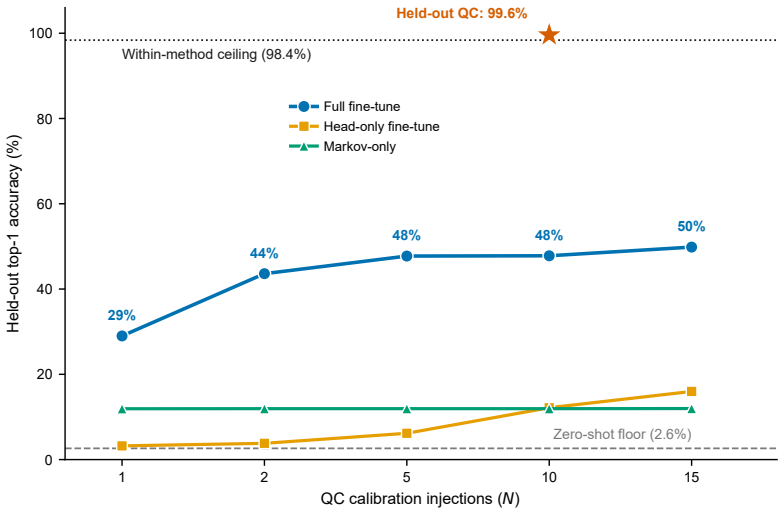
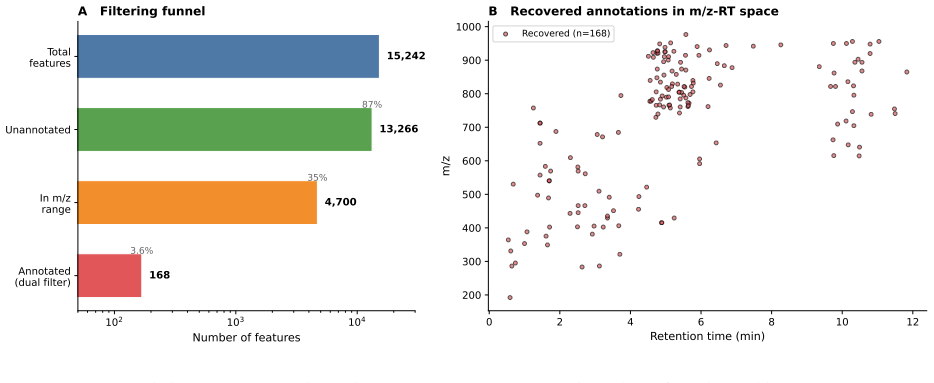
read the original abstract
Untargeted liquid chromatography-high-resolution mass spectrometry (LC-HRMS) detects thousands of molecular features per sample, yet only 2-20% receive confident structural annotations. A root cause of this "dark metabolome" is that tandem MS/MS acquisition is reactive: instruments select precursors only after ions appear, blind to what elutes next. We reframe chromatographic elution as an autoregressive sequence prediction task. Because reversed-phase elution order is governed by hydrophobicity, successive features form a physically constrained sequence, like tokens in language. We discretize the mass-to-charge (m/z) axis into 110 bins and train long short-term memory (LSTM) and Transformer models to predict the next eluting m/z bin from five annotation-free per-token features: m/z bin, mass defect, retention-time gap, polarity, and intensity rank. Trained on 15,242 features from four clinical lipidomics cohorts (342 plasma samples; SCIEX TripleTOF 6600+, Waters CSH C18), the LSTM reaches 98.4% top-1 accuracy (99.99% top-5; mean absolute error 3.6 Da) and the Transformer 98.0%. Ablation shows autoregressive context accounts for 55.5 percentage points while no single feature contributes more than 0.2 pp: the sequential pattern, not molecular properties, drives prediction. Models transfer across instruments sharing the method (r=0.999 on an independent Agilent 6530 dataset) but fail under a different column chemistry (5.1% top-1) or polarity mode (2.6%), confirming method- and mode-specificity. Fine-tuning on as few as two to five quality-control injections recovers held-out accuracy from 2.6% to nearly 50%, so cross-condition deployment needs minimal calibration. These results establish that elution sequences are highly predictable and lay the groundwork for predictive MS/MS acquisition to improve annotation coverage in untargeted metabolomics.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper reframes untargeted LC-HRMS lipidomics elution as an autoregressive sequence prediction task. It discretizes m/z into 110 bins and trains LSTM and Transformer models on five annotation-free per-feature statistics (m/z bin, mass defect, RT gap, polarity, intensity rank) from 15,242 features across 342 plasma samples. The LSTM achieves 98.4% top-1 accuracy (99.99% top-5, MAE 3.6 Da) on held-out data; ablation attributes 55.5 pp to autoregressive context. Models transfer across instruments with the same method (r=0.999) but not column chemistry or polarity, and fine-tuning on 2-5 QC injections recovers performance. The work aims to enable predictive MS/MS acquisition.
Significance. If the central empirical result holds under causal feature definitions, the high accuracies, the dominance of sequential context over individual features, the method-specific transfer, and the minimal-calibration fine-tuning results would be notable strengths. They provide concrete evidence that elution order is highly predictable from annotation-free statistics and directly support the motivating application of proactive MS/MS triggering to increase annotation coverage. The transfer and few-shot adaptation experiments are particularly useful for practical deployment.
major comments (2)
- [Abstract and Results (feature definition and ablation)] Abstract (feature list) and Results (ablation and accuracy claims): Intensity rank is defined as a feature's position in the global sorted intensity list of all detected features in the sample. In the autoregressive regime required for predictive MS/MS (predicting the (k+1)th feature after observing only the first k), the ranks of the observed features are not yet determined because later-eluting features may have higher intensities and reorder the ranking. This renders the feature non-causal and unavailable at inference time. The reported 98.4% top-1 accuracy, 99.99% top-5, and the 55.5 pp ablation gain for autoregressive context therefore rest on a statistic that cannot be computed in the online setting the paper targets. The other four features are causal, but the inclusion of intensity rank undermines the claim that the sequence can be learned from annotation-free per-feature statist
- [Methods and Results (transfer and ablation)] Methods (model training and evaluation) and Results (transfer tests): The paper reports strong transfer (r=0.999) only across instruments sharing the identical reversed-phase method and failure (5.1% and 2.6% top-1) under changed column chemistry or polarity. While this correctly demonstrates method specificity, the evaluation does not include a controlled test of the model under strictly causal feature computation (i.e., intensity ranks computed only from features observed so far). Such a test is required to establish whether the high accuracies survive removal or causal approximation of the non-causal feature.
minor comments (1)
- [Methods] The discretization into exactly 110 m/z bins and the precise definition of mass defect and RT gap should be stated with explicit formulas or bin boundaries in the Methods section for reproducibility.
Simulated Author's Rebuttal
We thank the referee for their thorough review and for pointing out the critical issue with the causality of the intensity rank feature in our autoregressive model. This comment is well-taken and directly impacts the applicability to the predictive MS/MS use case. We address the points below and will revise the manuscript accordingly.
read point-by-point responses
-
Referee: Abstract (feature list) and Results (ablation and accuracy claims): Intensity rank is defined as a feature's position in the global sorted intensity list of all detected features in the sample. In the autoregressive regime required for predictive MS/MS (predicting the (k+1)th feature after observing only the first k), the ranks of the observed features are not yet determined because later-eluting features may have higher intensities and reorder the ranking. This renders the feature non-causal and unavailable at inference time. The reported 98.4% top-1 accuracy, 99.99% top-5, and the 55.5 pp ablation gain for autoregressive context therefore rest on a statistic that cannot be computed in the online setting the paper targets. The other four features are causal, but the inclusion of intensity rank undermines the claim that the sequence can be learned from annotation-free per-feature statist
Authors: We agree with this assessment. The global intensity rank is indeed non-causal for online prediction. However, our ablation analysis indicates that individual features contribute minimally (≤0.2 pp), with the autoregressive context providing the majority of the predictive power (55.5 pp). We will revise the manuscript to remove intensity rank from the feature set and re-train/evaluate the models to demonstrate that performance is largely preserved. Additionally, we will include an ablation using a causal intensity rank (computed only from features observed up to the current point) to further validate the results under strictly causal conditions. These changes will be reflected in the revised version. revision: yes
-
Referee: Methods (model training and evaluation) and Results (transfer tests): The paper reports strong transfer (r=0.999) only across instruments sharing the identical reversed-phase method and failure (5.1% and 2.6% top-1) under changed column chemistry or polarity. While this correctly demonstrates method specificity, the evaluation does not include a controlled test of the model under strictly causal feature computation (i.e., intensity ranks computed only from features observed so far). Such a test is required to establish whether the high accuracies survive removal or causal approximation of the non-causal feature.
Authors: We acknowledge that the reported transfer and ablation results were obtained using the original non-causal feature set. To address this, we will perform additional experiments in the revised manuscript where intensity rank is either omitted or replaced with its causal counterpart. We will report accuracies, ablations, and transfer performance under these causal feature definitions to confirm the robustness of our findings. This will directly test whether the central empirical results hold without the non-causal feature. revision: yes
Circularity Check
No circularity; empirical ML evaluation on held-out data is self-contained
full rationale
The paper reports LSTM/Transformer accuracies (98.4% top-1) and ablations directly from training and testing on held-out features extracted from real LC-HRMS runs. No equations, parameter fits, or self-citations are used to derive the accuracy numbers; they are measured outcomes on unseen sequences. The five input features are computed from the data and supplied to the model; the reported performance is therefore an external empirical result rather than a reduction to the inputs by construction. Transfer tests on an independent instrument further provide an external check. The intensity-rank feature may limit online applicability, but that is a correctness or deployment issue, not a circularity in the derivation chain.
Axiom & Free-Parameter Ledger
free parameters (2)
- m/z bin count
- context length
axioms (2)
- domain assumption Reversed-phase elution order is governed by hydrophobicity and forms a physically constrained sequence.
- domain assumption Annotation-free per-token features suffice to capture the sequential pattern.
Reference graph
Works this paper leans on
-
[1]
Oswald Quehenberger, Aaron M Armando, Alex H Brown, et al. Lipidomics reveals a remarkable diversity of lipids in human plasma.J Lipid Res, 51(11):3299–3305, 2010. doi: 10.1194/jlr.M009449
-
[2]
Understanding the diversity of membrane lipid composition
Takeshi Harayama and Howard Riezman. Understanding the diversity of membrane lipid composition. Nat Rev Mol Cell Biol, 19(5):281–296, 2018. doi: 10.1038/nrm.2017.138
-
[3]
The mystery of membrane organization: composition, regulation and roles of lipid rafts.Nat Rev Mol Cell Biol, 18(6):361–374,
Erdinc Sezgin, Ilya Levental, Satyajit Mayor, and Christian Eggeling. The mystery of membrane organization: composition, regulation and roles of lipid rafts.Nat Rev Mol Cell Biol, 18(6):361–374,
-
[4]
doi: 10.1038/nrm.2017.16
-
[5]
James A. Olzmann and Pedro Carvalho. Dynamics and functions of lipid droplets.Nat Rev Mol Cell Biol, 20(3):137–155, 2019. doi: 10.1038/s41580-018-0085-z
-
[6]
Edward A. Dennis and Paul C. Norris. Eicosanoid storm in infection and inflammation.Nat Rev Immunol, 15(8):511–523, 2015. doi: 10.1038/nri3859
-
[7]
Yusuf A. Hannun and Lina M. Obeid. Sphingolipids and their metabolism in physiology and disease. Nat Rev Mol Cell Biol, 19(3):175–191, 2018. doi: 10.1038/nrm.2017.107
-
[8]
Matthias P. Wymann and Roger Schneiter. Lipid signalling in disease.Nat Rev Mol Cell Biol, 9(2): 162–176, 2008. doi: 10.1038/nrm2335
-
[9]
Peter Libby, Julie E. Buring, Lina Badimon, Göran K. Hansson, John Deanfield, Márcio S. Bittencourt, Lale Tokgözo˘glu, and Eldrin F. Lewis. Atherosclerosis.Nat Rev Dis Primers, 5(1):56, 2019. doi: 10.1038/s41572-019-0106-z
-
[10]
Todd A. Lydic and Young-Hwa Goo. Lipidomics unveils the complexity of the lipidome in metabolic diseases.Clinical and Translational Medicine, 7, 2018. doi: 10.1186/s40169-018-0182-9. URL https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5786598/
-
[11]
Lipidomes in health and dis- ease: Analytical strategies and considerations.TrAC Trends in Analytical Chemistry, 120:115664,
Fang Wei, Santosh Lamichhane, Matej Oreši ˇc, and Tuulia Hyötyläinen. Lipidomes in health and dis- ease: Analytical strategies and considerations.TrAC Trends in Analytical Chemistry, 120:115664,
-
[12]
URLhttps://www.sciencedirect.com/science/ article/pii/S0165993619303723
doi: 10.1016/j.trac.2019.115664. URLhttps://www.sciencedirect.com/science/ article/pii/S0165993619303723
-
[13]
Tomas Cajka, Jennifer T. Smilowitz, and Oliver Fiehn. Validating quantitative untargeted lipidomics across nine liquid chromatography–high-resolution mass spectrometry platforms.Anal Chem, 89(22): 12360–12368, 2017. doi: 10.1021/acs.analchem.7b03404
-
[14]
Patti, Oscar Yanes, and Gary Siuzdak
Gary J. Patti, Oscar Yanes, and Gary Siuzdak. Metabolomics: the apogee of the omic trilogy.Nat Rev Mol Cell Biol, 13(4):263–269, 2012. doi: 10.1038/nrm3314
-
[15]
Oliver Fiehn and Jayoung Kim. Metabolomics insights into pathophysiological mechanisms of inter- stitial cystitis.International Neurourology Journal, 18(3):106–114, 2014. doi: 10.5213/inj.2014.18.3
-
[16]
URLhttps://www.ncbi.nlm.nih.gov/pmc/articles/PMC4180160/
-
[17]
Schymanski, Junho Jeon, Rebekka Gulde, Kathrin Fenner, Martin Ruff, Heinz P
Emma L. Schymanski, Junho Jeon, Rebekka Gulde, Kathrin Fenner, Martin Ruff, Heinz P. Singer, and Juliane Hollender. Identifying small molecules via high resolution mass spectrometry: Communicating confidence.Environmental Science & Technology, 48(4):2097–2098, 2014. doi: 10.1021/es5002105
-
[18]
Zhiwei Zhou, Mingdu Luo, Haosong Zhang, Yandong Yin, Yuping Cai, and Zheng-Jiang Zhu. Metabo- lite annotation from knowns to unknowns through knowledge-guided multi-layer metabolic network- 32 ing.Nature Communications, 13(1), 2022. doi: 10.1038/s41467-022-34537-6
-
[19]
Neural networks and physical systems with emergent collective com- putational abilities
Ricardo R. da Silva, Pieter C. Dorrestein, and Robert A. Quinn. Illuminating the dark matter in metabolomics.Proc Natl Acad Sci U S A, 112(41):12549–12550, 2015. doi: 10.1073/pnas. 1516878112
-
[20]
María Eugenia Monge, James N. Dodds, Erin S. Baker, Arthur S. Edison, and Facundo M. Fernández. Challenges in identifying the dark molecules of life.Annu Rev Anal Chem, 12(1):177–199, 2019. doi: 10.1146/annurev-anchem-061318-114959
-
[21]
Ihab Hajjar, Chang Liu, Dean P. Jones, and Karan Uppal. Untargeted metabolomics reveal dysregula- tions in sugar, methionine, and tyrosine pathways in the prodromal state of AD.Alzheimers Dement (Amst), 12(1):e12064, 2020. doi: 10.1002/dad2.12064
-
[22]
Li Chen, Wenyun Lu, Lin Wang, et al. Metabolite discovery through global annotation of untargeted metabolomics data.Nat Methods, 18(11):1377–1385, 2021. doi: 10.1038/s41592-021-01303-3
-
[23]
Mingxun Wang, Jeremy J. Carver, Vanessa V . Phelan, Laura M. Sanchez, Neha Garg, Yao Peng, et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking.Nat Biotechnol, 34(8):828–837, 2016. doi: 10.1038/nbt.3597
-
[24]
Hisayuki Horai, Masanori Arita, Shigehiko Kanaya, et al. MassBank: a public repository for sharing mass spectral data for life sciences.J Mass Spectrom, 45(7):703–714, 2010. doi: 10.1002/jms.1777
-
[25]
SIRIUS 4: a rapid tool for turning tandem mass spectra into metabolite structure information.Nat Methods, 16(4):299–302, 2019
Kai Dührkop, Markus Fleischauer, Marcus Ludwig, et al. SIRIUS 4: a rapid tool for turning tandem mass spectra into metabolite structure information.Nat Methods, 16(4):299–302, 2019. doi: 10.1038/ s41592-019-0344-8
2019
-
[26]
Kai Dührkop, Louis-Félix Nothias, Markus Fleischauer, et al. Systematic classification of unknown metabolites using high-resolution fragmentation mass spectra.Nat Biotechnol, 39(4):462–471, 2021. doi: 10.1038/s41587-020-0740-8
-
[27]
Florian Huber, Sven van der Burg, Justin J. J. van der Hooft, and Lars Ridder. MS2DeepScore: a novel deep learning similarity measure to compare tandem mass spectra.J Cheminform, 13(1):84, 2021. doi: 10.1186/s13321-021-00558-4
-
[28]
Felicity Allen, Allison Pon, Michael Wilson, Russ Greiner, and David Wishart. CFM-ID: a web server for annotation, spectrum prediction and metabolite identification from tandem mass spectra.Nucleic Acids Res, 42(W1):W94–W99, 2014. doi: 10.1093/nar/gku436
-
[29]
Paolo Bonini, Tobias Kind, Hiroshi Tsugawa, Dinesh Kumar Barupal, and Oliver Fiehn. Retip: Re- tention time prediction for compound annotation in untargeted metabolomics.Analytical Chemistry, 92(11):7515–7522, 2020. doi: 10.1021/acs.analchem.9b05765. URLhttps://pubs.acs.org/doi/ 10.1021/acs.analchem.9b05765
-
[30]
Robin Schmid, Steffen Heuckeroth, Ansgar Korf, et al. RT-Transformer: retention time prediction for metabolite annotation to assist in metabolite identification.Bioinformatics, 40(3):btae084, 2024. doi: 10.1093/bioinformatics/btae084
-
[31]
Michael Witting and Sebastian Böcker. Current status of retention time prediction in metabolite identi- fication.Journal of Separation Science, 43(9-10):1746–1754, 2020. doi: https://doi.org/10.1002/jssc. 202000060. URLhttps://onlinelibrary.wiley.com/doi/abs/10.1002/jssc.202000060. 33
-
[32]
Eric Bach, Emma L. Schymanski, and Juho Rousu. Joint structural annotation of small molecules using liquid chromatography retention order and tandem mass spectrometry data.Nature Machine Intelligence, 4(12):1224–1237, 2022. doi: 10.1038/s42256-022-00577-2
-
[33]
Fang-Yuan Sun, Ying-Hao Yin, Hui-Jun Liu, Lu-Na Shen, Xiu-Lin Kang, Gui-Zhong Xin, Li-Fang Liu, and Jia-Yi Zheng. Roasmi: accelerating small molecule identification by repurposing retention data.Journal of Cheminformatics, 17(1), 2025. doi: 10.1186/s13321-025-00968-8
-
[34]
Emmanuel Defossez, Julien Bourquin, Stephan von Reuss, Sergio Rasmann, and Gaétan Glauser. Eight key rules for successful data-dependent acquisition in mass spectrometry-based metabolomics.Mass Spectrometry Reviews, 42(1):131–143, 2021. doi: 10.1002/mas.21715
-
[35]
Hanane El Boudlali, Laura Lehmicke, and Uta Ceglarek. High-resolution accurate mass- mass spec- trometry based- untargeted metabolomics: Reproducibility and detection power across data-dependent acquisition, data-independent acquisition, and acquirex.Computational and Structural Biotechnology Journal, 27:2412–2423, 2025. doi: 10.1016/j.csbj.2025.05.046
-
[36]
Brianna T. Cooper, Ruohong Yang, et al. An assessment of acquirex and compound discoverer software 3.3 for non-targeted metabolomics.Scientific Reports, 2024. doi: 10.1038/s41598-024-55356-3
-
[37]
Joe Wandy, Vinny Davies, Ross McBride, Stefan Weidt, Simon Rogers, and Rónán Daly. Vimms 2.0: A framework to develop, test and optimise fragmentation strategies in lc-ms metabolomics.Journal of Open Source Software, 7(71):3990, 2022. doi: 10.21105/joss.03990
-
[38]
Kyowon Jeong, Author Corresponding, et al. Flashida enables intelligent data acquisition for top-down proteomics to boost proteoform identification counts.Nature Communications, 13:4407, 2022. doi: 10.1038/s41467-022-31922-z
-
[39]
Christoph Wichmann, Florian Meier, Sebastian Virreira Winter, Andreas-David Brunner, Jürgen Cox, and Matthias Mann. Maxquant.live enables global targeting of more than 25,000 peptides.Molecular & Cellular Proteomics, 18(5):982a–994, 2019. doi: 10.1074/mcp.tir118.001131
-
[40]
Barshop, Seema Sharma, Jesse Canterbury, Aaron M
Brandon Bills, William D. Barshop, Seema Sharma, Jesse Canterbury, Aaron M. Robitaille, Michael Goodwin, Michael W. Senko, and Vlad Zabrouskov. Novel real-time library search driven data ac- quisition strategy for identification and characterization of metabolites.Analytical Chemistry, 94(9): 3749–3755, 2022. doi: 10.1021/acs.analchem.1c04336
-
[41]
Spatially Resolving Electron Spin Resonance ofπ-Radical in Single-Molecule Magnet
Lilian R. Heil, Philip M. Remes, Jesse D. Canterbury, Ping Yip, William D. Barshop, Christine C. Wu, and Michael J. MacCoss. Dynamic data-independent acquisition mass spectrometry with real- time retrospective alignment.Analytical Chemistry, 95(32):11854–11858, 2023. doi: 10.1021/acs. analchem.3c00903
work page doi:10.1021/acs 2023
-
[42]
Jake B. White, Paul J. Trim, Thalia Salagaras, Aaron Long, Peter J. Psaltis, Johan W. Verjans, and Marten F. Snel. Equivalent carbon number and interclass retention time conversion enhance lipid identification in untargeted clinical lipidomics.Analytical Chemistry, 94(8):3476–3484, 2022. doi: 10.1021/acs.analchem.1c03770
-
[43]
Retention dependences support highly confident identification of lipid species in human plasma by reversed-phase uhplc/ms.Analytical and Bioanalytical Chemistry, 414(1):319–331, 34
Zuzana Va ˇnková, Ond ˇrej Peterka, Michaela Chocholoušková, Denise Wolrab, Robert Jirásko, and Michal Hol ˇcapek. Retention dependences support highly confident identification of lipid species in human plasma by reversed-phase uhplc/ms.Analytical and Bioanalytical Chemistry, 414(1):319–331, 34
-
[44]
doi: 10.1007/s00216-021-03492-4
-
[45]
Eric Bach, Sandor Szedmak, Céline Brouard, Shenghuo Liang, and Juho Rousu. Liquid- chromatography retention order prediction for metabolite identification.Bioinformatics, 34(17):i875– i883, 2018. doi: 10.1093/bioinformatics/bty590
-
[46]
Jan Stanstrup, Steffen Neumann, and Urska Vrhošek. PredRet: Prediction of retention time by direct mapping between multiple chromatographic systems.Analytical Chemistry, 87(18):9421–9428, 2015. doi: 10.1021/acs.analchem.5b02287
-
[47]
Álvaro González-Domínguez, Núria Estanyol-Torres, Carl Brunius, Rikard Landberg, and Raúl González-Domínguez. Qcomics: Recommendations and guidelines for robust, easily implementable and reportable quality control of metabolomics data.Analytical Chemistry, 96(3):1064–1072, 2024. doi: 10.1021/acs.analchem.3c03660
-
[48]
Ashish Vaswani, Noam Shazeer, Niki Parmar, Jakob Uszkoreit, Llion Jones, Aidan N Gomez, Łukasz Kaiser, and Illia Polosukhin. Attention is all you need. InAdvances in Neural Information Processing Systems, volume 30, pages 6000–6010, 2017. doi: 10.48550/arXiv.1706.03762
work page internal anchor Pith review Pith/arXiv arXiv doi:10.48550/arxiv.1706.03762 2017
-
[49]
Lawrence Zitnick, Jerry Ma, and Rob Fergus
Alexander Rives, Joshua Meier, Tom Sercu, Siddharth Goyal, Zeming Lin, Jason Liu, Demi Guo, Myle Ott, C. Lawrence Zitnick, Jerry Ma, and Rob Fergus. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences.Proc Natl Acad Sci U S A, 118(15): e2016239118, 2021. doi: 10.1073/pnas.2016239118
-
[50]
Hunter, Costas Bekas, and Alpha A
Philippe Schwaller, Teodoro Laino, Théophile Gaudin, Peter Bolgar, Christopher A. Hunter, Costas Bekas, and Alpha A. Lee. Molecular Transformer: A model for uncertainty-calibrated chemical reac- tion prediction.ACS Cent Sci, 5(9):1572–1583, 2019. doi: 10.1021/acscentsci.9b00576
-
[51]
Lewis, Yicheng Jin, Xiuyu Tang, Vidit Shah, Christina Doty, Bethany E
Nicholas R. Lewis, Yicheng Jin, Xiuyu Tang, Vidit Shah, Christina Doty, Bethany E. Matthews, Sarah Akers, and Steven R. Spurgeon. Forecasting of in situ electron energy loss spectroscopy.npj Compu- tational Materials, 8(1), 2022. doi: 10.1038/s41524-022-00940-2
-
[52]
Roman Bushuiev, Anton Bushuiev, Raman Samusevich, Corinna Brungs, Josef Sivic, and Tomáš Pluskal. Self-supervised learning of molecular representations from millions of tandem mass spec- tra using DreaMS.Nat Biotechnol, 2025. doi: 10.1038/s41587-025-02663-3. Online ahead of print
-
[53]
Campbell, Jack Geremia, and Timothy Kassis
Gabriel Asher, Mimoun Cadosh Delmar, Jennifer M. Campbell, Jack Geremia, and Timothy Kassis. LSM1-MS2: A foundation model for MS/MS, encompassing chemical property predictions, search and de novo generation, 2024. ChemRxiv preprint
2024
-
[54]
Leon L. Xu and Hannes L. Röst. Peak detection on data independent acquisition mass spectrometry data with semisupervised convolutional transformers, 2020. arXiv:2010.13841
arXiv 2020
-
[55]
Hiroshi Tsugawa, Tomas Cajka, Tobias Kind, Yan Ma, Brendan Higgins, Kazutaka Ikeda, Mitsuhiro Kanazawa, Jean VanderGheynst, Oliver Fiehn, and Masanori Arita. MS-DIAL: data-independent MS/MS deconvolution for comprehensive metabolome analysis.Nat Methods, 12(6):523–526, 2015. doi: 10.1038/nmeth.3393
-
[56]
Interleukin-1 blockade in recently decompensated systolic heart failure: Results from REDHART.Circ Heart Fail, 10(11):e004373,
Benjamin W Van Tassell, Justin M Canada, Salvatore Carbone, et al. Interleukin-1 blockade in recently decompensated systolic heart failure: Results from REDHART.Circ Heart Fail, 10(11):e004373,
-
[57]
doi: 10.1161/CIRCHEARTFAILURE.117.004373. 35
-
[58]
Metabolic modulation predicts heart failure tests performance.PLoS One, 14(6):e0218153, 2019
Daniel Jr Contaifer, Leo F Buckley, George Wohlford, Naren Gajenthra Kumar, Joshua M Morriss, Asanga D Ranasinghe, Salvatore Carbone, Justin M Canada, Cory Trankle, Antonio Abbate, Ben- jamin W Van Tassell, and Dayanjan S Wijesinghe. Metabolic modulation predicts heart failure tests performance.PLoS One, 14(6):e0218153, 2019. doi: 10.1371/journal.pone.0218153
-
[59]
Daniel Jr Contaifer, Catherine H Roberts, Naren Gajenthra Kumar, Ramesh Natarajan, others, and Dayanjan S Wijesinghe. A preliminary investigation towards the risk stratification of allogeneic stem cell recipients with respect to the potential for development of GVHD via their pre-transplant plasma lipid and metabolic signature.Cancers (Basel), 11(4):466, ...
-
[60]
Sara Martínez, Miguel Fernández-García, Sara Londoño-Osorio, Coral Barbas, and Ana Gradillas. Highly reliable LC-MS lipidomics database for efficient human plasma profiling based on NIST SRM 1950.J Lipid Res, 65(11):100671, 2024. doi: 10.1016/j.jlr.2024.100671
-
[61]
Harmonizing lipidomics: NIST interlaboratory comparison exercise for lipidomics using SRM 1950-metabolites in frozen human plasma.J Lipid Res, 58(12):2275–2288,
John A Bowden, Alan Heckert, Candice Z Ulmer, Christina M Jones, Jeremy P Koelmel, Laila Ab- dullah, Linda Ahonen, et al. Harmonizing lipidomics: NIST interlaboratory comparison exercise for lipidomics using SRM 1950-metabolites in frozen human plasma.J Lipid Res, 58(12):2275–2288,
1950
-
[62]
doi: 10.1194/jlr.M079012
-
[63]
Reinke, Julia Kuligowski, Ian D
David Broadhurst, Royston Goodacre, Stacey N. Reinke, Julia Kuligowski, Ian D. Wilson, Matthew R. Lewis, and Warwick B. Dunn. Guidelines and considerations for the use of system suitability and quality control samples in mass spectrometry assays applied in untargeted clinical metabolomic stud- ies.Metabolomics, 14(5):72, 2018. doi: 10.1007/s11306-018-1367-3
-
[64]
Paul Benton, Julijana Ivanisevic, Nathaniel G
H. Paul Benton, Julijana Ivanisevic, Nathaniel G. Mahieu, Michael E. Kurczy, Caroline H. Johnson, Lauren Franco, Duane Rinehart, Elizabeth Valentine, Harsha Gowda, Baljit K. Ubhi, Ralf Tautenhahn, Andrew Gieschen, Matthew W. Fields, Gary J. Patti, and Gary Siuzdak. Autonomous metabolomics for rapid metabolite identification in global profiling.Analytical ...
-
[65]
Randolph, Matthew Muhoberac, Palak Manchanda, and Gaurav Chopra
Connor Beveridge, Sanjay Iyer, Caitlin E. Randolph, Matthew Muhoberac, Palak Manchanda, and Gaurav Chopra. Claw-mrm: Comprehensive lipidomics automation workflow for multiple reaction monitoring using large language models.Analytical Chemistry, 2025. doi: 10.1021/acs.analchem. 4c05039
-
[66]
Gustav K. Reder, E. Yves Bjurstrom, Daniel Brunnsaker, Fanny Kronstrom, Paul Lasin, Ievgeniia Tiukova, Otto I. Savolainen, James N. Dodds, Jody C. May, John P. Wikswo, John A. McLean, and Ross D. King. Autonoms: Automated ion mobility metabolomic fingerprinting.Journal of the Ameri- can Society for Mass Spectrometry, 35(3):542–550, 2024. doi: 10.1021/jasm...
-
[67]
Self-supervised learning from small-molecule mass spectrometry data.Nat Biotechnol, 2025
Wout Bittremieux and William Stafford Noble. Self-supervised learning from small-molecule mass spectrometry data.Nat Biotechnol, 2025. doi: 10.1038/s41587-025-02677-x. Online ahead of print
-
[68]
Language model-guided anticipation and discovery of mammalian metabolites.Nature, 2026
Hantao Qiang, Fei Wang, Wenyun Lu, Xi Xing, Hahn Kim, et al. Language model-guided anticipation and discovery of mammalian metabolites.Nature, 2026. doi: 10.1038/s41586-025-09969-x. 36
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.