Single-Cell Cross-Modal Transfer by Adversarial Fine-Tuning of Foundation Models
Pith reviewed 2026-06-27 22:20 UTC · model grok-4.3
The pith
A single-cell foundation model translates between unpaired spatial transcriptomics and scRNA-seq data via adversarial fine-tuning.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Adversarial fine-tuning of a single-cell foundation model enables cross-modal translation between unpaired ST and scRNA-seq datasets, recovering information about former in situ neighbourhoods that whole-transcriptome readouts are known to retain.
What carries the argument
Adversarial fine-tuning of a single-cell foundation model to achieve unpaired cross-modal transfer between ST and scRNA-seq.
If this is right
- The method works without paired individual cells or spots, allowing use of separately collected datasets.
- It outperforms dedicated multi-omics translation methods on the translation task.
- Spatial neighbourhood information can be imputed onto scRNA-seq profiles from available ST references.
- Both modalities can be leveraged in their abundant unpaired forms rather than waiting for matched samples.
Where Pith is reading between the lines
- The same fine-tuning strategy could be tested on additional single-cell layers such as chromatin accessibility or protein measurements.
- Applying the model across many tissue types would test whether neighbourhood retention is a general property of scRNA-seq data.
- The approach suggests foundation models may serve as flexible starting points for other unpaired modality transfers in single-cell work.
Load-bearing premise
Whole-transcriptome readouts from dissociated scRNA-seq cells retain recoverable information about their original spatial neighbourhoods.
What would settle it
On paired ST-scRNA-seq test data, the translated profiles would show no better spatial alignment or neighbourhood recovery than a random or non-adversarial baseline.
Figures
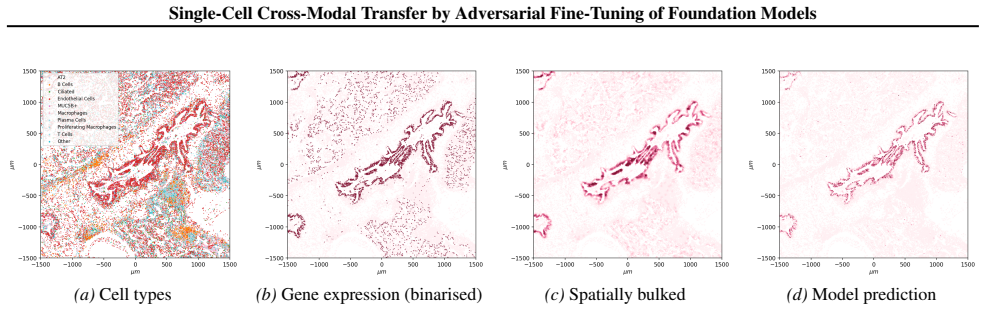
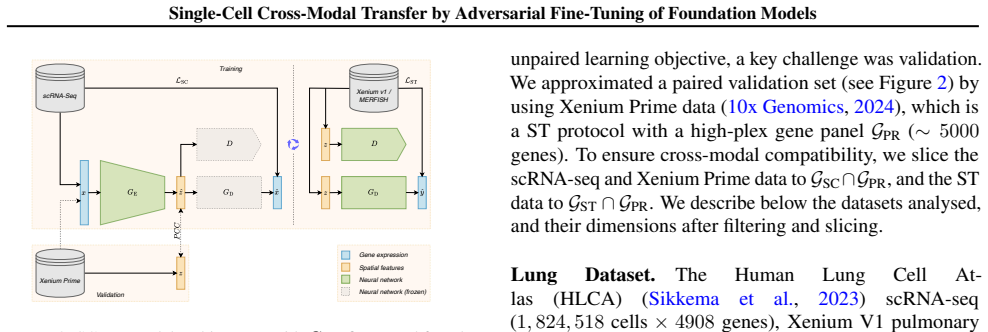
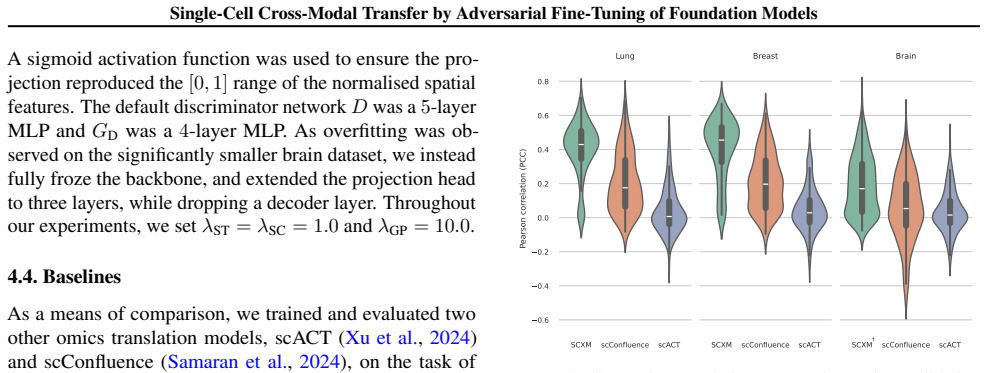
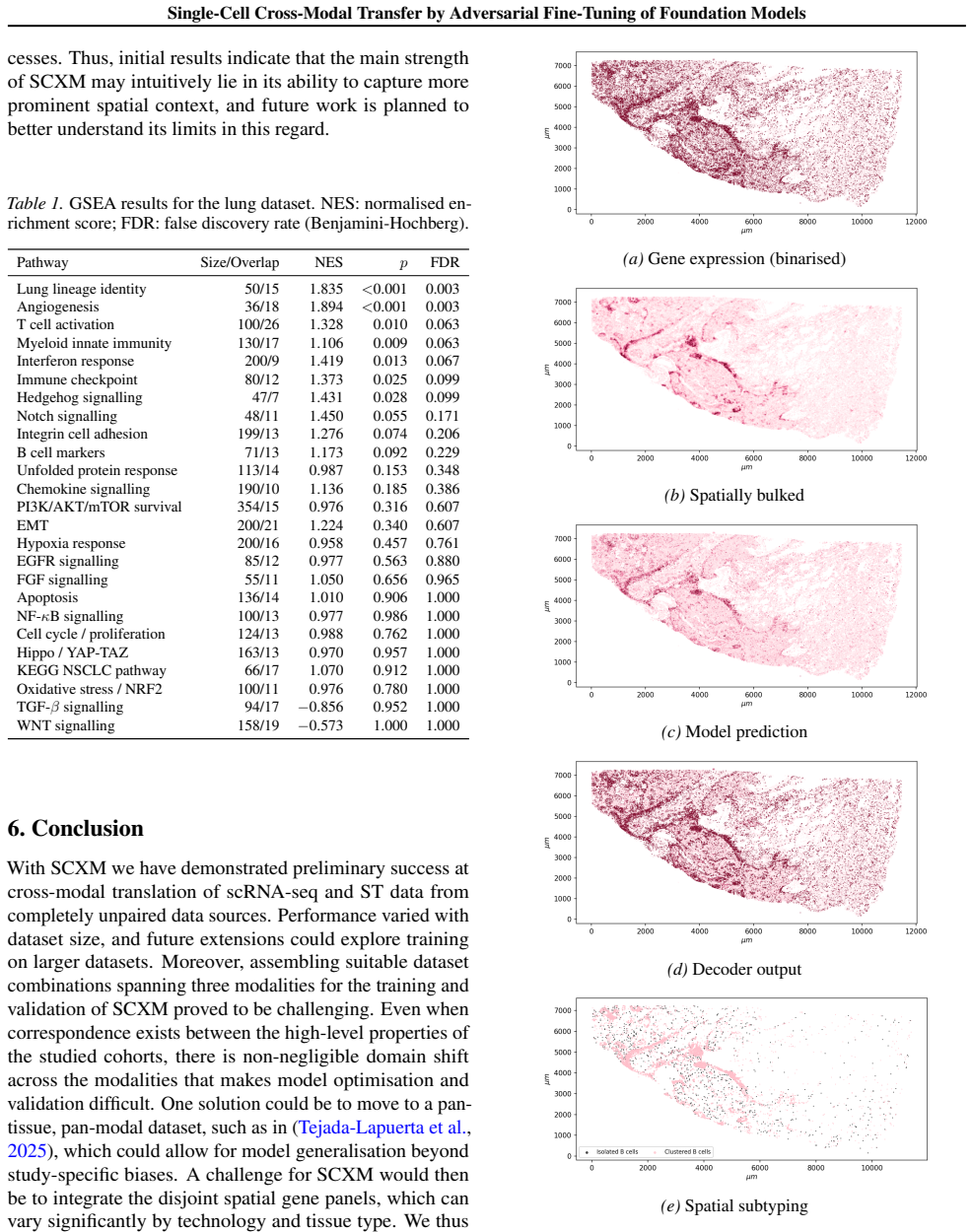
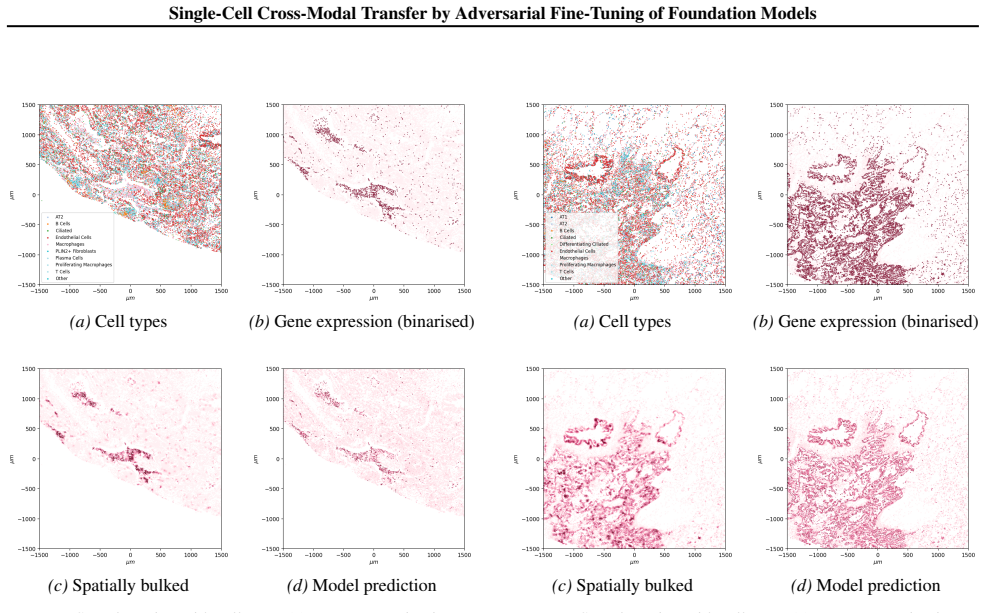
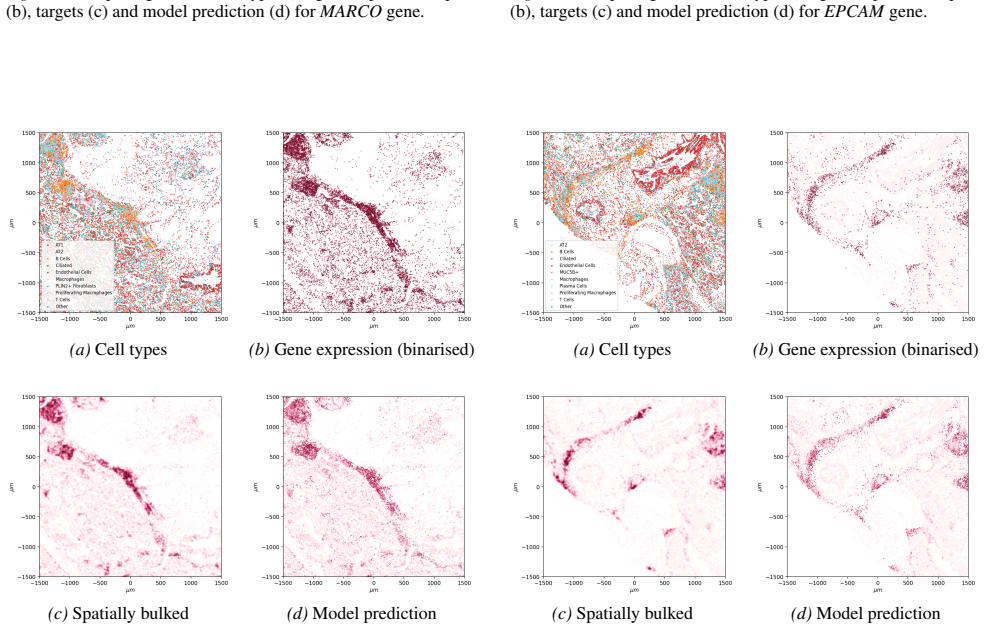
read the original abstract
Spatial transcriptomics (ST) is a powerful tool for exploring biological properties dependent on structure, proximity, and interaction in tissue. The methods underpinning ST are developing rapidly but are limited in their ability to profile many thousands of genes at a subcellular scale. Although dissociated from tissue, it is known that the whole-transcriptome readouts of cells in single-cell RNA sequencing (scRNA-seq) retain information about their former in situ neighbourhoods, motivating computational methods to recover it. While paired ST and scRNA-seq datasets are scarce, each modality in its own right is abundantly available. We therefore propose to perform cross-modal translation between unpaired ST and scRNA-seq data. In this work we show that a single-cell foundation model can perform this translation via adversarial fine-tuning. We demonstrate that our method performs favourably against methods built for multi-omics translation.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper proposes performing cross-modal translation between unpaired spatial transcriptomics (ST) and single-cell RNA-seq (scRNA-seq) datasets by adversarially fine-tuning a single-cell foundation model. It claims this yields favorable performance relative to methods designed for multi-omics translation, motivated by the premise that scRNA-seq readouts retain information about cells' original in situ neighborhoods.
Significance. If the central claim holds after proper validation, the work could enable spatial inference from the large existing corpus of unpaired scRNA-seq data, reducing reliance on scarce paired ST-scRNA-seq collections. The adversarial fine-tuning of foundation models is a relevant technical direction, but significance is limited by the absence of quantified evidence on signal retention strength or detailed performance metrics in the available description.
major comments (1)
- [Abstract] Abstract: The statement that 'it is known that the whole-transcriptome readouts of cells in single-cell RNA sequencing (scRNA-seq) retain information about their former in situ neighbourhoods' is presented as established without apparent quantification of signal strength, noise level, or context dependence. This retention is the load-bearing premise for the claim that adversarial fine-tuning can recover spatial neighborhoods; if the retained signal is weak or absent, the method cannot create information that is not present.
Simulated Author's Rebuttal
We thank the referee for their review and for highlighting the foundational premise of our work. We address the major comment below and will incorporate revisions to strengthen the presentation of this point.
read point-by-point responses
-
Referee: [Abstract] Abstract: The statement that 'it is known that the whole-transcriptome readouts of cells in single-cell RNA sequencing (scRNA-seq) retain information about their former in situ neighbourhoods' is presented as established without apparent quantification of signal strength, noise level, or context dependence. This retention is the load-bearing premise for the claim that adversarial fine-tuning can recover spatial neighborhoods; if the retained signal is weak or absent, the method cannot create information that is not present.
Authors: We agree that the abstract states the premise without explicit quantification of signal strength or context dependence. The full manuscript motivates this claim from established observations in the spatial transcriptomics literature that microenvironmental effects produce detectable correlations between gene expression and spatial position even after dissociation. Our empirical results provide indirect quantification by showing that adversarial fine-tuning of the foundation model yields superior cross-modal translation performance relative to multi-omics baselines; this outcome would not be possible if the retained spatial signal were absent or too weak to exploit. We will revise the abstract to reference this empirical support and to note that the method's success serves as a functional test of signal usability, while adding a short discussion of known context dependence (e.g., tissue type and cell density) drawn from the results section. revision: yes
Circularity Check
No circularity: method proposal rests on external premise treated as known, with empirical validation
full rationale
The paper proposes an adversarial fine-tuning approach for cross-modal translation between unpaired ST and scRNA-seq data using a single-cell foundation model. The abstract presents the key premise ('it is known that the whole-transcriptome readouts of cells in single-cell RNA sequencing (scRNA-seq) retain information about their former in situ neighbourhoods') as established background rather than deriving it. No equations, fitted parameters renamed as predictions, self-citations forming load-bearing uniqueness claims, or ansatzes smuggled via prior work are present in the provided text. The central claim is empirical (favourable performance vs. multi-omics baselines), which is self-contained against external benchmarks and does not reduce to its own inputs by construction.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption whole-transcriptome readouts of cells in scRNA-seq retain information about their former in situ neighbourhoods
Reference graph
Works this paper leans on
-
[1]
Nature communications , volume =
High resolution mapping of the tumor microenvironment using integrated single-cell, spatial and in situ analysis , author =. Nature communications , volume =. 2023 , publisher =
2023
-
[2]
Nucleic acids research , volume =
CZ CELLxGENE Discover: a single-cell data platform for scalable exploration, analysis and modeling of aggregated data , author =. Nucleic acids research , volume =. 2025 , publisher =
2025
-
[3]
Nature genetics , volume =
Spatial transcriptomics identifies molecular niche dysregulation associated with distal lung remodeling in pulmonary fibrosis , author =. Nature genetics , volume =. 2025 , publisher =
2025
-
[4]
Nature medicine , volume =
An integrated cell atlas of the lung in health and disease , author =. Nature medicine , volume =. 2023 , publisher =
2023
-
[5]
Statistical genomics: methods and protocols , pages =
The gene expression omnibus database , author =. Statistical genomics: methods and protocols , pages =. 2016 , publisher =
2016
-
[6]
Cell , volume =
Molecular and spatial signatures of mouse brain aging at single-cell resolution , author =. Cell , volume =. 2023 , publisher =
2023
-
[7]
NAR Genomics and Bioinformatics , volume =
A highly resolved integrated single-cell atlas of human breast cancers , author =. NAR Genomics and Bioinformatics , volume =. 2026 , publisher =
2026
-
[8]
bioRxiv , pages =
Biomarker Quantification in Breast Cancer using Xenium In Situ , author =. bioRxiv , pages =. 2025 , publisher =
2025
-
[9]
Post-Xenium In Situ Applications: Immunofluorescence, H&E, Visium v2, and Visium HD , author =
-
[10]
Mouse Brain Nuclei Isolated with Chromium Nuclei Isolation Kit, SaltyEZ Protocol, and 10x Complex Tissue DP (CT Sorted and CT Unsorted) , author =
-
[11]
Xenium In Situ Gene Expression - Post-Xenium Analyzer H&E Staining , author =
-
[12]
Nature , volume =
Transfer learning enables predictions in network biology , author =. Nature , volume =. 2023 , publisher =
2023
-
[13]
Nature methods , volume =
SpaGCN: Integrating gene expression, spatial location and histology to identify spatial domains and spatially variable genes by graph convolutional network , author =. Nature methods , volume =. 2021 , publisher =
2021
-
[14]
Nature Genetics , volume =
Quantitative characterization of cell niches in spatially resolved omics data , author =. Nature Genetics , volume =. 2025 , publisher =
2025
-
[15]
biorxiv , pages =
scGPT-spatial: Continual pretraining of single-cell foundation model for spatial transcriptomics , author =. biorxiv , pages =. 2025 , publisher =
2025
-
[16]
Nature Communications , volume =
scConfluence: single-cell diagonal integration with regularized Inverse Optimal Transport on weakly connected features , author =. Nature Communications , volume =. 2024 , publisher =
2024
-
[17]
Proceedings of the 33rd ACM International Conference on Information and Knowledge Management , pages =
scACT: accurate cross-modality translation via cycle-consistent training from unpaired single-cell data , author =. Proceedings of the 33rd ACM International Conference on Information and Knowledge Management , pages =
-
[18]
Briefings in Bioinformatics , volume =
scDCT: a conditional diffusion-based deep learning model for high-fidelity single-cell cross-modality translation , author =. Briefings in Bioinformatics , volume =. 2025 , publisher =
2025
-
[19]
Genome Biology , volume =
scDOT: optimal transport for mapping senescent cells in spatial transcriptomics , author =. Genome Biology , volume =. 2024 , publisher =
2024
-
[20]
Nature methods , pages =
Nicheformer: a foundation model for single-cell and spatial omics , author =. Nature methods , pages =. 2025 , publisher =
2025
-
[21]
Nature machine intelligence , volume =
scBERT as a large-scale pretrained deep language model for cell type annotation of single-cell RNA-seq data , author =. Nature machine intelligence , volume =. 2022 , publisher =
2022
-
[22]
Nature Communications , year =
scLong: A billion-parameter foundation model for capturing long-range gene context in single-cell transcriptomics , author =. Nature Communications , year =
-
[23]
Patterns , volume =
CellContrast: Reconstructing spatial relationships in single-cell RNA sequencing data via deep contrastive learning , author =. Patterns , volume =. 2024 , publisher =
2024
-
[24]
BMC genomics , volume =
BioMart--biological queries made easy , author =. BMC genomics , volume =. 2009 , publisher =
2009
-
[25]
Nature methods , volume =
Squidpy: a scalable framework for spatial omics analysis , author =. Nature methods , volume =. 2022 , publisher =
2022
-
[26]
bioRxiv , year =
Quantized multi-task learning for context-specific representations of gene network dynamics , author =. bioRxiv , year =
-
[27]
Biometrika , volume =
Notes on continuous stochastic phenomena , author =. Biometrika , volume =. 1950 , publisher =
1950
-
[28]
The incorporated statistician , volume =
The contiguity ratio and statistical mapping , author =. The incorporated statistician , volume =. 1954 , publisher =
1954
-
[29]
arXiv preprint arXiv:1511.05644 , year =
Adversarial autoencoders , author =. arXiv preprint arXiv:1511.05644 , year =
-
[30]
Nature Reviews Genetics , volume =
Deciphering cell--cell interactions and communication from gene expression , author =. Nature Reviews Genetics , volume =. 2021 , publisher =
2021
-
[31]
Proceedings of the IEEE international conference on computer vision , pages =
Unpaired image-to-image translation using cycle-consistent adversarial networks , author =. Proceedings of the IEEE international conference on computer vision , pages =
-
[32]
Advances in Neural Information Processing Systems , volume =
Schrodinger bridge flow for unpaired data translation , author =. Advances in Neural Information Processing Systems , volume =
-
[33]
arXiv preprint arXiv:2104.05358 , year =
Unit-ddpm: Unpaired image translation with denoising diffusion probabilistic models , author =. arXiv preprint arXiv:2104.05358 , year =
-
[34]
International conference on machine learning , pages =
Wasserstein generative adversarial networks , author =. International conference on machine learning , pages =. 2017 , organization =
2017
-
[35]
Advances in neural information processing systems , volume =
Improved training of wasserstein gans , author =. Advances in neural information processing systems , volume =
-
[36]
Communications of the ACM , volume =
Generative adversarial networks , author =. Communications of the ACM , volume =. 2020 , publisher =
2020
-
[37]
Proceedings of the IEEE/CVF conference on computer vision and pattern recognition , pages =
Exploring simple siamese representation learning , author =. Proceedings of the IEEE/CVF conference on computer vision and pattern recognition , pages =
-
[38]
BioRxiv , pages =
Scaling large language models for next-generation single-cell analysis , author =. BioRxiv , pages =
-
[39]
European conference on computer vision , pages =
Contrastive learning for unpaired image-to-image translation , author =. European conference on computer vision , pages =. 2020 , organization =
2020
-
[40]
Nature methods , volume =
scGPT: toward building a foundation model for single-cell multi-omics using generative AI , author =. Nature methods , volume =. 2024 , publisher =
2024
-
[41]
Bert: Pre-training of deep bidirectional transformers for language understanding , author =. Proceedings of the 2019 conference of the North American chapter of the association for computational linguistics: human language technologies, volume 1 (long and short papers) , pages =
2019
-
[42]
Nature methods , volume =
Large-scale foundation model on single-cell transcriptomics , author =. Nature methods , volume =. 2024 , publisher =
2024
-
[43]
Genome biology , volume =
SCANPY: large-scale single-cell gene expression data analysis , author =. Genome biology , volume =. 2018 , publisher =
2018
-
[44]
Science , volume =
Spatially resolved, highly multiplexed RNA profiling in single cells , author =. Science , volume =. 2015 , publisher =
2015
-
[45]
The EMBO journal , volume =
The RING finger protein Siah-1 regulates the level of the transcriptional coactivator OBF-1 , author =. The EMBO journal , volume =. 2001 , publisher =
2001
-
[46]
Frontiers in immunology , volume =
The role of macrophage scavenger receptor 1 (MSR1) in inflammatory disorders and cancer , author =. Frontiers in immunology , volume =. 2022 , publisher =
2022
-
[47]
Proceedings of the National Academy of Sciences , volume =
Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles , author =. Proceedings of the National Academy of Sciences , volume =. 2005 , publisher =
2005
-
[48]
Science , volume =
Cross-tissue immune cell analysis reveals tissue-specific features in humans , author =. Science , volume =. 2022 , publisher =
2022
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.