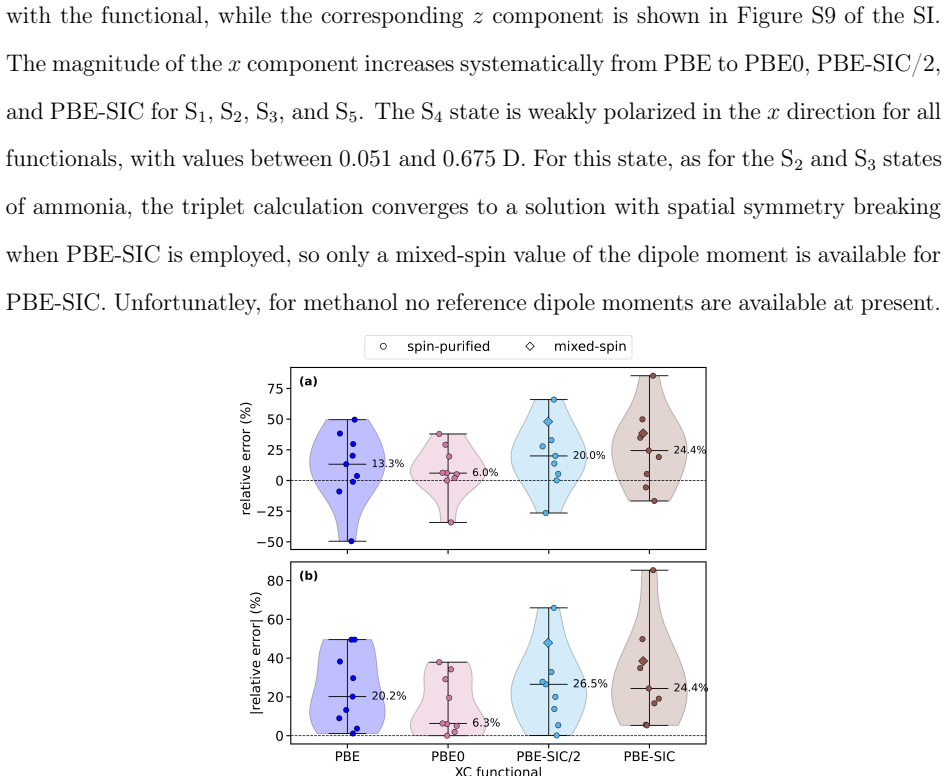
Excited-state Properties Beyond the Excitation Energy from Orbital-Optimized Density Functional Calculations I: Dipole Moments of Rydberg States
Pith reviewed 2026-06-27 08:00 UTC · model grok-4.3
The pith
Orbital-optimized DFT with plane waves reveals that atomic basis sets produce inaccurate dipole moments for Rydberg states even when excitation energies are correct.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
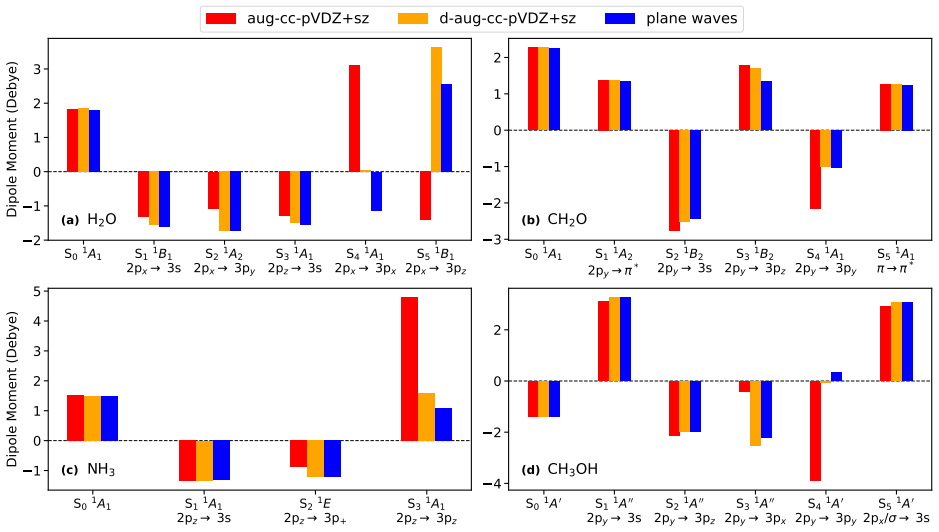
Orbital-optimized density functional calculations with a plane waves basis set are used to compute the dipole moments of several Rydberg states of a set of molecules. Plane waves provide a flexible representation of the diffuse Rydberg orbitals, revealing limitations of atomic orbitals basis sets. A commonly used single-augmented atomic basis set yields inaccurate dipole moments even when the excitation energy is insensitive to the basis representation, and discrepancies with plane waves calculations persist for the most diffuse states even when extra augmented diffuse functions are added. The generalized gradient approximation functional PBE gives good agreement with higher-level calculatio
What carries the argument
Orbital-optimized density functional calculations with a plane-waves basis set used to represent diffuse Rydberg orbitals when evaluating dipole moments.
If this is right
- A commonly used single-augmented atomic basis set yields inaccurate dipole moments even when the excitation energy is insensitive to the basis representation.
- Discrepancies with plane waves calculations persist for the most diffuse states even when extra augmented diffuse functions are added.
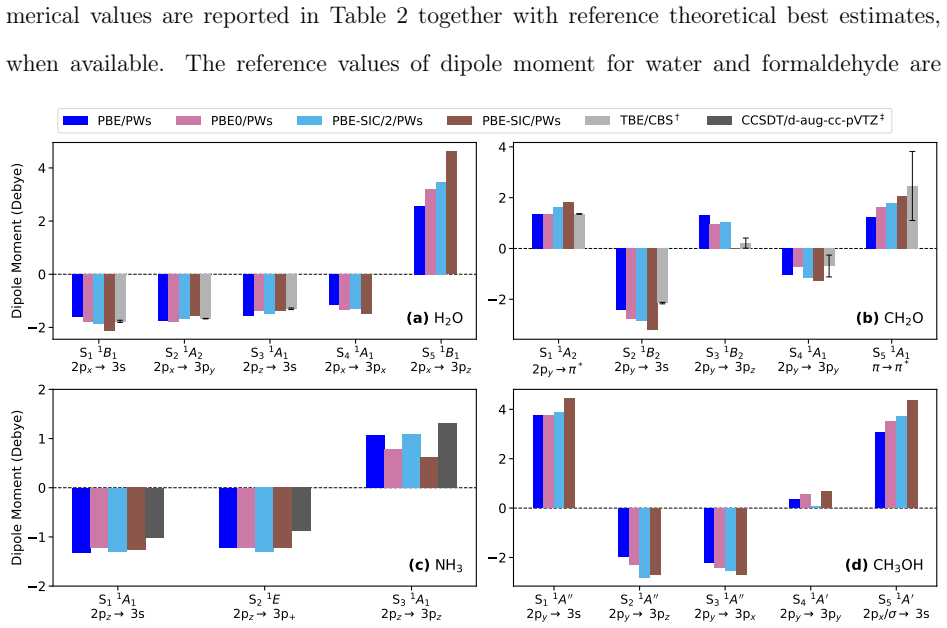
- The PBE functional gives good agreement with higher-level calculations for the dipole moments.
- The hybrid functional PBE0 further improves the dipole-moment results.
- PBE with globally scaled explicit Perdew-Zunger self-interaction correction leads to larger errors and overestimation of the dipole moment.
Where Pith is reading between the lines
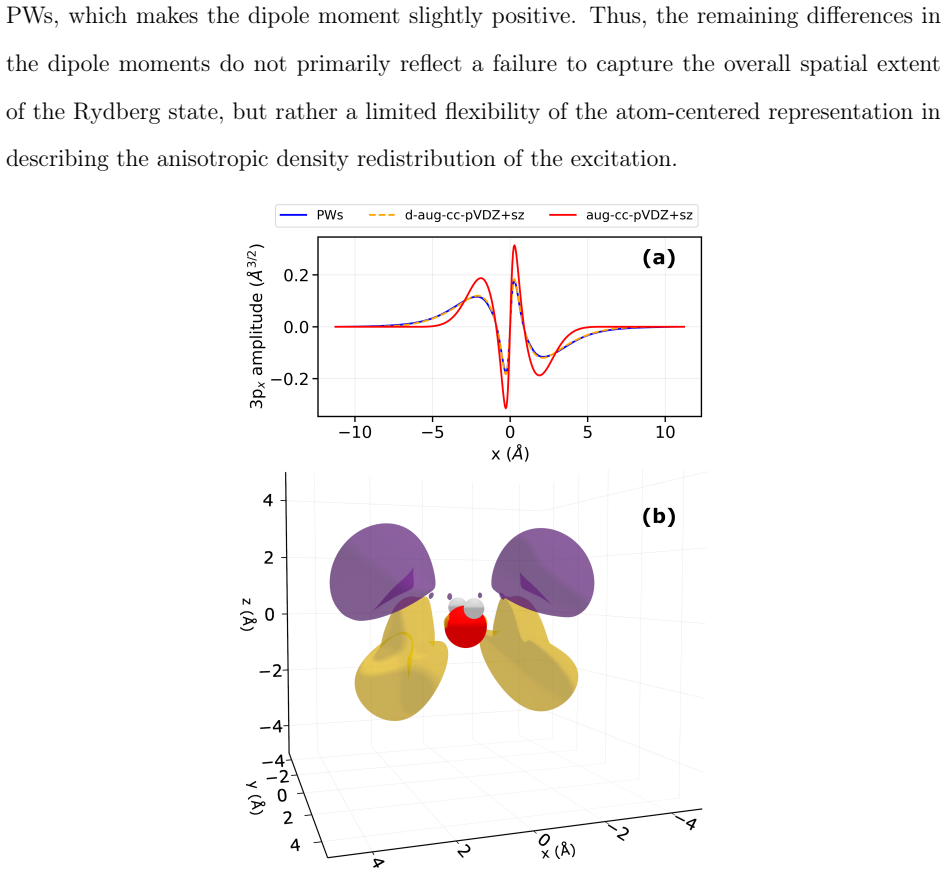
- Properties other than energy may require more flexible basis representations for Rydberg states than what suffices for energies alone.
- The same basis limitations could appear in other diffuse-state observables such as transition dipole moments or polarizabilities.
- Plane-wave OO-DFT could be tested on additional molecular excited-state properties to check whether the pattern holds.
Load-bearing premise
The plane-wave calculations with the chosen cutoff and k-point sampling form a converged reference against which atomic-basis errors can be measured.
What would settle it
A new plane-wave run with substantially higher cutoff energy or denser k-point grid that changes the computed dipole moments by more than the reported discrepancies would falsify the reference.
Figures
read the original abstract
Rydberg excited states are challenging to describe due to their highly diffuse character. Orbital-optimized density functional calculations provide a better description of Rydberg states than time-dependent density functional theory. However, benchmarks have so far focused on the excitation energy, while assessments of dipole moments remain limited to the lowest excited state. Here, orbital-optimized density functional calculations with a plane waves basis set are used to compute the dipole moments of several Rydberg states of a set of molecules. Plane waves provide a flexible representation of the diffuse Rydberg orbitals, revealing limitations of atomic orbitals basis sets. A commonly used single-augmented atomic basis set yields inaccurate dipole moments even when the excitation energy is insensitive to the basis representation, and discrepancies with plane waves calculations persist for the most diffuse states even when extra augmented diffuse functions are added. The generalized gradient approximation functional PBE gives good agreement with higher-level calculations where available. The hybrid functional PBE0 further improves the results, while PBE with globally scaled explicit Perdew-Zunger self-interaction correction leads to larger errors and an overestimation of the dipole moment, despite restoring the correct asymptotic $-1/r$ behavior of the effective Kohn--Sham potential.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript uses orbital-optimized DFT calculations in a plane-wave basis to compute dipole moments of multiple Rydberg excited states across a set of molecules. It reports that commonly employed single-augmented atomic basis sets produce inaccurate dipoles even when excitation energies are insensitive to basis choice, that adding further diffuse functions does not eliminate discrepancies for the most diffuse states, and that PBE and PBE0 yield good agreement with higher-level references while PBE with Perdew-Zunger SIC overestimates the dipoles despite restoring the correct asymptotic potential.
Significance. If the central comparisons hold, the work is significant because it isolates a clear limitation of standard atomic bases for excited-state properties (dipoles) that is not apparent from excitation energies alone, and it demonstrates the utility of plane-wave representations for diffuse orbitals. The systematic functional comparison (PBE, PBE0, SIC-PBE) against external higher-level data is a positive feature.
major comments (2)
- [Methods] The headline claim that single-augmented atomic bases yield inaccurate dipoles (while energies remain stable) and that extra diffuse functions still fail for the most diffuse states (abstract) rests on the plane-wave results constituting a converged reference. However, the Methods section provides no dipole-specific convergence tests (varying cutoff at fixed geometry or increasing k-point density) for the extended Rydberg densities; residual PW incompleteness would produce errors of the same character as the atomic-basis errors being criticized.
- [Abstract / Results] The abstract states that PBE gives good agreement with higher-level calculations 'where available' and that PBE0 further improves results, yet no quantitative error statistics, specific molecules, or tabulated deviations with error bars are referenced in the summary of results; without these, the strength of the functional-performance conclusion cannot be assessed.
minor comments (2)
- [Abstract] The abstract refers to 'a set of molecules' without naming them or indicating the number of states per molecule; adding this information would improve readability.
- Notation for the self-interaction correction (explicitly 'globally scaled explicit Perdew-Zunger') should be defined at first use with a brief equation or reference.
Simulated Author's Rebuttal
We thank the referee for their thoughtful review and for recognizing the significance of our findings on the limitations of atomic basis sets for Rydberg dipole moments. We address the major comments below and will revise the manuscript accordingly.
read point-by-point responses
-
Referee: [Methods] The headline claim that single-augmented atomic bases yield inaccurate dipoles (while energies remain stable) and that extra diffuse functions still fail for the most diffuse states (abstract) rests on the plane-wave results constituting a converged reference. However, the Methods section provides no dipole-specific convergence tests (varying cutoff at fixed geometry or increasing k-point density) for the extended Rydberg densities; residual PW incompleteness would produce errors of the same character as the atomic-basis errors being criticized.
Authors: We agree that demonstrating convergence of the plane-wave dipole moments is essential to support our claims. Although our calculations used a high plane-wave cutoff and Gamma-point sampling with sufficiently large simulation cells, we did not explicitly report dipole convergence tests in the Methods. In the revised manuscript, we will include additional convergence data for the dipole moments with respect to cutoff energy and cell size for representative Rydberg states to confirm that the reported values are converged to within acceptable tolerances. revision: yes
-
Referee: [Abstract / Results] The abstract states that PBE gives good agreement with higher-level calculations 'where available' and that PBE0 further improves results, yet no quantitative error statistics, specific molecules, or tabulated deviations with error bars are referenced in the summary of results; without these, the strength of the functional-performance conclusion cannot be assessed.
Authors: The full results section of the manuscript does contain quantitative comparisons, including tables with dipole moments from different methods and deviations from reference values for specific molecules. However, to make this clearer in the summary, we will add a sentence in the results section or abstract referencing the mean absolute deviations or specific error ranges (e.g., PBE0 errors typically below 0.1 D for available cases). We will ensure error bars or statistics are explicitly highlighted. revision: partial
Circularity Check
No circularity: direct computational benchmarks against external references
full rationale
The paper computes dipole moments of Rydberg states via orbital-optimized DFT in a plane-wave basis and directly compares the results to atomic-orbital basis sets and to independent higher-level calculations where available. No derivation chain reduces a reported quantity to a fitted parameter or self-citation by construction; the central claims rest on explicit numerical comparisons rather than on any of the enumerated circular patterns. The plane-wave results function as an external reference, not as a quantity derived from the atomic-basis data under test.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption Orbital-optimized Kohn-Sham DFT provides a valid description of Rydberg excited states
- domain assumption Plane-wave basis sets can be converged to serve as reference for diffuse orbitals
Forward citations
Cited by 2 Pith papers
-
Excited-state Properties Beyond the Excitation Energy from Orbital-Optimized Density Functional Calculations II: Absorption Spectra
Orbital-optimized DFT with extended Löwdin formalism qualitatively reproduces multireference absorption spectra for single-determinant states but shows discrepancies for multi-configurational ones, with no systematic ...
-
Orbital-optimized density functional calculations of excited electronic states: Recent advances and perspectives
Review summarizing theoretical foundations, recent algorithmic advances, open-shell singlet treatments, transition properties, and applications of orbital-optimized DFT to Rydberg, charge-transfer, and core excitations.
Reference graph
Works this paper leans on
-
[1]
Sarkar, Rudraditya and Boggio-Pasqua, Martial and Loos, Pierre-François and Jacquemin, Denis. Benchmarking TD-DFT and wave function methods for oscillator strengths and excited-state dipole moments. J. Chem. Theory Comput. doi:10.1021/acs.jctc.0c01228
-
[2]
Chutjian, A and Hall, R I and Trajmar, S. Electron-impact excitation of H2O and D2O at various scattering angles and impact energies in the energy-loss range 4.2–12 eV. J. Chem. Phys. doi:10.1063/1.431370
-
[3]
Chrayteh, Amara and Blondel, Aymeric and Loos, Pierre-François and Jacquemin, Denis. Mountaineering strategy to excited states: Highly accurate oscillator strengths and dipole moments of small molecules. J. Chem. Theory Comput. doi:10.1021/acs.jctc.0c01111
-
[4]
A theoretical determination of the electronic spectrum of formaldehyde
Merchán, Manuela and Roos, Björn O. A theoretical determination of the electronic spectrum of formaldehyde. Theoret. Chim. Acta. doi:10.1007/bf01125948
-
[5]
Excited states of the water molecule: analysis of the valence and Rydberg character
Rubio, Mercedes and Serrano-Andrés, Luis and Merchán, Manuela. Excited states of the water molecule: analysis of the valence and Rydberg character. J. Chem. Phys. doi:10.1063/1.2837827
-
[6]
Quantum Theory of Many-Particle Systems
Löwdin, Per-Olov. Quantum Theory of Many-Particle Systems. I. Physical Interpretations by Means of Density Matrices, Natural Spin-Orbitals, and Convergence Problems in the Method of Configurational Interaction. Phys. Rev. doi:10.1103/PhysRev.97.1474
-
[7]
Figari, Giuseppe and Magnasco, Valerio. On the evaluation of the cofactors occurring in the matrix elements between multiply-excited determinantal wavefunctions of non-orthogonal orbitals. Mol. Phys. doi:10.1080/00268978500101351
-
[8]
Generalized gradient approximation made simple
Perdew, J P and Burke, K and Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. doi:10.1103/PhysRevLett.77.3865
-
[9]
Generalized Gradient Approximation Made Simple [Phys
Perdew, John P and Burke, Kieron and Ernzerhof, Matthias. Generalized Gradient Approximation Made Simple [Phys. Rev. Lett. 77, 3865 (1996)]. Phys. Rev. Lett. doi:10.1103/PhysRevLett.78.1396
-
[11]
Projector augmented-wave method
Blöchl, P E. Projector augmented-wave method. Phys. Rev. B Condens. Matter. doi:10.1103/physrevb.50.17953
-
[12]
Electron affinities of the first-row atoms revisited
Kendall, Rick A and Dunning, Thom H and Harrison, Robert J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. doi:10.1063/1.462569
-
[13]
A New Basis Set Exchange: An Open, Up-to-date Resource for the Molecular Sciences Community
Pritchard, Benjamin P and Altarawy, Doaa and Didier, Brett and Gibsom, Tara D and Windus, Theresa L. A New Basis Set Exchange: An Open, Up-to-date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. doi:10.1021/acs.jcim.9b00725
-
[14]
Gaussian basis sets for use in correlated molecular calculations
Woon, David E and Dunning, Thom H. Gaussian basis sets for use in correlated molecular calculations. IV. Calculation of static electrical response properties. J. Chem. Phys. doi:10.1063/1.466439
-
[15]
Gaussian basis sets for use in correlated molecular calculations
Dunning, Thom H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. doi:10.1063/1.456153
-
[16]
Software update: The ORCA program system—version 6.0
Neese, Frank. Software update: The ORCA program system—version 6.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. doi:10.1002/wcms.70019
-
[17]
Neese, Frank. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. doi:10.1002/wcms.81
-
[18]
Time-dependent density functional theory within the Tamm–Dancoff approximation
Hirata, So and Head-Gordon, Martin. Time-dependent density functional theory within the Tamm–Dancoff approximation. Chem. Phys. Lett. doi:10.1016/S0009-2614(99)01149-5
-
[19]
Real-space grid implementation of the projector augmented wave method
Mortensen, J J and Hansen, L B and Jacobsen, K W. Real-space grid implementation of the projector augmented wave method. Phys. Rev. B. doi:10.1103/PhysRevB.71.035109
-
[20]
a fer, Christian and J \'o nsson, Elvar \
Mortensen, Jens J rgen and Larsen, Ask Hjorth and Kuisma, Mikael and Ivanov, Aleksei V and Taghizadeh, Alireza and Peterson, Andrew and Haldar, Anubhab and Dohn, Asmus Ougaard and Sch \"a fer, Christian and J \'o nsson, Elvar \"O rn and Hermes, Eric D and Nilsson, Fredrik Andreas and Kastlunger, Georg and Levi, Gianluca and J \'o nsson, Hannes and H \"a k...
-
[21]
Self-consistent field calculations of excited states using the maximum overlap method (MOM)
Gilbert, Andrew T B and Besley, Nicholas A and Gill, Peter M W. Self-consistent field calculations of excited states using the maximum overlap method (MOM). J. Phys. Chem. A. doi:10.1021/jp801738f
-
[22]
Hait, Diptarka and Head-Gordon, Martin. Highly accurate prediction of core spectra of molecules at density functional theory cost: Attaining sub-electronvolt error from a restricted open-shell Kohn-Sham approach. J. Phys. Chem. Lett. doi:10.1021/acs.jpclett.9b03661
-
[23]
Reliable transition properties from excited-state mean-field calculations
Bourne Worster, Susannah and Feighan, Oliver and Manby, Frederick R. Reliable transition properties from excited-state mean-field calculations. J. Chem. Phys. doi:10.1063/5.0041233
-
[24]
Highly accurate and robust constraint-based orbital-optimized core excitations
Lemke, Yannick and Kussmann, Jörg and Ochsenfeld, Christian. Highly accurate and robust constraint-based orbital-optimized core excitations. J. Phys. Chem. A. doi:10.1021/acs.jpca.4c04139
-
[25]
Optimization of Wave Function and Geometry in the Finite Basis Hartree-Fock Method , volume =
Martin Head-Gordon and John A Pople , issue =. Optimization of Wave Function and Geometry in the Finite Basis Hartree-Fock Method , volume =. J. Phys. Chem , pages =
-
[26]
Hait, Diptarka and Haugen, Eric A and Yang, Zheyue and Oosterbaan, Katherine J and Leone, Stephen R and Head-Gordon, Martin. Accurate prediction of core-level spectra of radicals at density functional theory cost via square gradient minimization and recoupling of mixed configurations. J. Chem. Phys. doi:10.1063/5.0018833
-
[27]
Selenius, Elli and Sigurdarson, Alec Elías and Schmerwitz, Yorick L A and Levi, Gianluca. Orbital-optimized versus time-dependent density functional calculations of intramolecular charge transfer excited states. J. Chem. Theory Comput. doi:10.1021/acs.jctc.3c01319
-
[28]
Faraday Discussions , volume=
Variational calculations of excited states via direct optimization of the orbitals in DFT , author=. Faraday Discussions , volume=. 2020 , publisher=
2020
-
[29]
Ivanov, Aleksei V. and Levi, Gianluca and J. J. Chem. Theory Comput. , month =. doi:10.1021/acs.jctc.1c00157 , issn =
-
[30]
Sigurdarson, Alec E and Schmerwitz, Yorick L A and Tveiten, Dagrún K V and Levi, Gianluca and Jónsson, Hannes. Orbital-optimized density functional calculations of molecular Rydberg excited states with real space grid representation and self-interaction correction. J. Chem. Phys. doi:10.1063/5.0179271
-
[31]
Variational density functional calculations of excited states via direct optimization
Levi, Gianluca and Ivanov, Aleksei V and Jónsson, Hannes. Variational density functional calculations of excited states via direct optimization. J. Chem. Theory Comput. doi:10.1021/acs.jctc.0c00597
-
[32]
Role of Rydberg states in the photochemical dynamics of ethylene
Mori, Toshifumi and Glover, William J and Schuurman, Michael S and Martinez, Todd J. Role of Rydberg states in the photochemical dynamics of ethylene. J. Phys. Chem. A. doi:10.1021/jp2097185
-
[33]
Rydberg states: sensitive probes of molecular structure
Kuthirummal, Narayanan and Weber, Peter M. Rydberg states: sensitive probes of molecular structure. Chem. Phys. Lett. doi:10.1016/j.cplett.2003.08.002
-
[34]
Molecules in high Rydberg states
Merkt, F. Molecules in high Rydberg states. Annu. Rev. Phys. Chem. doi:10.1146/annurev.physchem.48.1.675
-
[35]
The role of Rydberg states in spectroscopy and photochemistry: Low and high Rydberg states
Sándorfy, C. The role of Rydberg states in spectroscopy and photochemistry: Low and high Rydberg states. doi:10.1007/0-306-46938-3
-
[36]
Jochim, Bethany and Siemering, R and Zohrabi, M and Voznyuk, O and Mahowald, J B and Schmitz, D G and Betsch, K J and Berry, Ben and Severt, T and Kling, Nora G and Burwitz, T G and Carnes, K D and Kling, M F and Ben-Itzhak, I and Wells, E and de Vivie-Riedle, R. The importance of Rydberg orbitals in dissociative ionization of small hydrocarbon molecules ...
-
[37]
van Harrevelt, Rob and van Hemert, Marc C. Photodissociation of water. I. Electronic structure calculations for the excited states. J. Chem. Phys. doi:10.1063/1.481153
-
[38]
Applications of molecular Rydberg states in chemical dynamics and spectroscopy
Softley, T P. Applications of molecular Rydberg states in chemical dynamics and spectroscopy. Int. Rev. Phys. Chem. doi:10.1080/01442350310001652940
-
[39]
Feller, David and Peterson, Kirk A and Davidson, Ernest R. A systematic approach to vertically excited states of ethylene using configuration interaction and coupled cluster techniques. J. Chem. Phys. doi:10.1063/1.4894482
-
[40]
A mountaineering strategy to excited states: Highly accurate reference energies and benchmarks
Loos, Pierre-François and Scemama, Anthony and Blondel, Aymeric and Garniron, Yann and Caffarel, Michel and Jacquemin, Denis. A mountaineering strategy to excited states: Highly accurate reference energies and benchmarks. J. Chem. Theory Comput. doi:10.1021/acs.jctc.8b00406
-
[41]
Applications of time-dependent and time-independent density functional theory to Rydberg transitions
Seidu, Issaka and Krykunov, Mykhaylo and Ziegler, Tom. Applications of time-dependent and time-independent density functional theory to Rydberg transitions. J. Phys. Chem. A. doi:10.1021/jp5082802
-
[42]
Ammonia: the prototypical lone pair molecule
Bartlett, Rodney J and Del Bene, Janet E and Perera, S Ajith and Mattie, Reneépeloquin. Ammonia: the prototypical lone pair molecule. Theochem. doi:10.1016/s0166-1280(97)90277-3
-
[43]
Journal of Chemical Theory and Computation , pages =
Levi, Gianluca and Kroesbergen, Max and Thirion, Louis and Schmerwitz, Yorick L A and Elvar, O J and Bilous, Pavlo and Hansmann, Philipp and Hannes, J , doi =. Journal of Chemical Theory and Computation , pages =
-
[44]
Journal of Chemical Theory and Computation , pages =
Schmerwitz, Yorick L A and Selenius, Elli and Levi, Gianluca , doi =. Journal of Chemical Theory and Computation , pages =
-
[45]
Schmerwitz, Yorick L. A. and Levi, Gianluca and J. J. Chem. Theory Comput. , number =. doi:10.1021/acs.jctc.3c00178 , eprint =
-
[46]
General-model-space state-universal coupled-cluster method: excitation energies of water
Li, Xiangzhu and Paldus, Josef. General-model-space state-universal coupled-cluster method: excitation energies of water. Mol. Phys. doi:10.1080/00268970500416145
-
[47]
Ciofini, Ilaria and Adamo, Carlo. Accurate evaluation of valence and low-lying Rydberg states with standard time-dependent density functional theory. J. Phys. Chem. A. doi:10.1021/jp0722152
-
[48]
Universal Gaussian basis sets for an optimum representation of Rydberg and continuum wavefunctions
Kaufmann, K and Baumeister, W and Jungen, M. Universal Gaussian basis sets for an optimum representation of Rydberg and continuum wavefunctions. J. Phys. B At. Mol. Opt. Phys. doi:10.1088/0953-4075/22/14/007
-
[49]
Simultaneous calculation of Rydberg and valence excited states of formaldehyde
Müller, Thomas and Lischka, Hans. Simultaneous calculation of Rydberg and valence excited states of formaldehyde. Theor. Chem. Acc. doi:10.1007/s002140100286
-
[50]
Zobel, J Patrick and Nogueira, Juan J and González, Leticia. The IPEA dilemma in CASPT2. Chem. Sci. doi:10.1039/c6sc03759c
-
[51]
Automated active space selection with dipole moments
Kaufold, Benjamin W and Chintala, Nithin and Pandeya, Pratima and Dong, Sijia S. Automated active space selection with dipole moments. J. Chem. Theory Comput. doi:10.1021/acs.jctc.2c01128
-
[52]
Veryazov, Valera and Malmqvist, Per ke and Roos, Bj \"o rn O. How to select active space for multiconfigurational quantum chemistry?: Selection of Active Space for Multiconfigurational Methods. Int. J. Quantum Chem. doi:10.1002/qua.23068
-
[53]
Theoretical study of the absorption and emission spectra of indole in the gas phase and in a solvent
Serrano-Andrés, Luis and Roos, Björn O. Theoretical study of the absorption and emission spectra of indole in the gas phase and in a solvent. J. Am. Chem. Soc. doi:10.1021/ja952035i
-
[54]
Theoretical study of the electronic spectrum of imidazole
Serrano-Andrés, Luis and Fülscher, Markus P and Roos, Björn O and Merchán, Manuela. Theoretical study of the electronic spectrum of imidazole. J. Phys. Chem. doi:10.1021/jp952809h
-
[55]
Excited state dipole moments from SCF: a benchmark
Paetow, Lukas and Neugebauer, Johannes. Excited state dipole moments from SCF: a benchmark. Phys. Chem. Chem. Phys. 2025 , pages =. doi:10.1039/d5cp01695a
-
[56]
Rationale for mixing exact exchange with density functional approximations
Perdew, John P and Ernzerhof, Matthias and Burke, Kieron. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. doi:10.1063/1.472933
-
[57]
Self-interaction correction to density-functional approximations for many-electron systems
Perdew, J P and Zunger, Alex. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B Condens. Matter. doi:10.1103/physrevb.23.5048
-
[58]
Melander, Marko and J \'o nsson, Elvar \"O and Mortensen, Jens J and Vegge, Tejs and Garc \' a Lastra, Juan Maria. Implementation of constrained DFT for computing charge transfer rates within the projector augmented wave method. J. Chem. Theory Comput. doi:10.1021/acs.jctc.6b00815
-
[59]
Systematic optimization of long-range corrected hybrid density functionals
Chai, Jeng-Da and Head-Gordon, Martin. Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. doi:10.1063/1.2834918
-
[60]
Absolute photoabsorption cross-sections of methanol for terrestrial and astrophysical relevance
Lange, Emanuele and Lozano, Ana Isabel and Jones, Nykola C and Hoffmann, Søren Vrønning and Kumar, Sarvesh and Śmiałek, Małgorzata A and Duflot, Denis and Brunger, Michael J and Limão-Vieira, Paulo. Absolute photoabsorption cross-sections of methanol for terrestrial and astrophysical relevance. J. Phys. Chem. A. doi:10.1021/acs.jpca.0c06615
-
[61]
On the calculation of multiplet energies by the hartree-fock-slater method
Ziegler, Tom and Rauk, Arvi and Baerends, Evert J. On the calculation of multiplet energies by the hartree-fock-slater method. Theoret. Chim. Acta. doi:10.1007/bf00551551
-
[62]
Yang, Ke and Peverati, Roberto and Truhlar, Donald G and Valero, Rosendo. Density functional study of multiplicity-changing valence and Rydberg excitations of p-block elements: delta self-consistent field, collinear spin-flip time-dependent density functional theory (DFT), and conventional time-dependent DFT. J. Chem. Phys. doi:10.1063/1.3607312
-
[63]
Recent Advances In Density Functional Methods: (Part I) , pages=
Time-dependent density functional response theory for molecules , author=. Recent Advances In Density Functional Methods: (Part I) , pages=. 1995 , publisher=
1995
-
[64]
Physical review , volume=
Inhomogeneous electron gas , author=. Physical review , volume=. 1964 , publisher=
1964
-
[65]
K U , doi =
Runge, Erich and Gross, E. K U , doi =. Phys. Rev. Lett. , number =
-
[66]
Rydberg energies using excited state density functional theory , author=. J. Chem. Phys. , volume=. 2008 , publisher=
2008
-
[67]
and Benfield, Peter and Helgaker, Trygve and Tozer, David J
Peach, Michael J.G. and Benfield, Peter and Helgaker, Trygve and Tozer, David J. , doi =. Journal of Chemical Physics , number =
-
[68]
and Jamorski, Christine and Casida, Kim C
Casida, Mark E. and Jamorski, Christine and Casida, Kim C. and Salahub, Dennis R. , doi =. Journal of Chemical Physics , number =
-
[69]
Journal of the American Chemical Society , number =
Shu, Yinan and Truhlar, Donald G , doi =. Journal of the American Chemical Society , number =
-
[70]
Buenker, Robert J and Peyerimhoff, Sigrid D. Mixed valence—Rydberg states. Chem. Phys. Lett. doi:10.1016/0009-2614(75)80271-5
-
[71]
Toffoli, Daniele and Quarin, Matteo and Fronzoni, Giovanna and Stener, Mauro. Accurate vertical excitation energies of BODIPY/Aza-BODIPY derivatives from excited-state mean-field calculations. J. Phys. Chem. A. doi:10.1021/acs.jpca.2c04473
-
[72]
Vandaele, Eva and Mališ, Momir and Luber, Sandra. The photodissociation of solvated cyclopropanone and its hydrate explored via non-adiabatic molecular dynamics using SCF. Phys. Chem. Chem. Phys. doi:10.1039/d1cp05187c
-
[73]
Borges, Jr, Itamar. Configuration interaction oscillator strengths of the H2O molecule: Transitions from the ground to the B ^1 A _1 , C ^1 1B _1 , D ^1 1A _1 , and 11B2 excited states. Chem. Phys. doi:10.1016/j.chemphys.2006.07.007
-
[74]
Ab initio study of valence and Rydberg states of CH3Br
Escure, Christelle and Leininger, Thierry and Lepetit, Bruno. Ab initio study of valence and Rydberg states of CH3Br. J. Chem. Phys. doi:10.1063/1.3152865
-
[75]
Zheng, Lianjun and Polizzi, Nicholas F and Dave, Adarsh R and Migliore, Agostino and Beratan, David N. Where is the electronic oscillator strength? Mapping oscillator strength across molecular absorption spectra. J. Phys. Chem. A. doi:10.1021/acs.jpca.6b00692
-
[76]
Reisler, Hanna and Krylov, Anna I. Interacting Rydberg and valence states in radicals and molecules: experimental and theoretical studies. Int. Rev. Phys. Chem. doi:10.1080/01442350902989170
-
[77]
doi:10.1021/ct500727c , issn =
Journal of Chemical Theory and Computation , number =. doi:10.1021/ct500727c , issn =
-
[78]
Lacombe, Lionel and Maitra, Neepa T. , doi =. npj Computational Materials , number =. 2023 , pages =. arXiv , arxivId =:2302.11366 , issn =
arXiv 2023
-
[79]
An ab initio exciton model including charge-transfer excited states
Li, Xin and Parrish, Robert M and Liu, Fang and Kokkila Schumacher, Sara I L and Martínez, Todd J. An ab initio exciton model including charge-transfer excited states. J. Chem. Theory Comput. doi:10.1021/acs.jctc.7b00171
-
[80]
On the determination of excitation energies using density functional theory
Tozer, David J and Handy, Nicholas C. On the determination of excitation energies using density functional theory. Phys. Chem. Chem. Phys. doi:10.1039/a910321j
-
[81]
Palmer, Michael H and Gordon, Agnieszka J. The electronic states of isothiazole studied by VUV absorption spectroscopy and ab initio configuration interaction methods. Chem. Phys. doi:10.1016/j.chemphys.2007.09.044
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.