Orbital-optimized density functional calculations of excited electronic states: Recent advances and perspectives
Pith reviewed 2026-06-27 04:59 UTC · model grok-4.3
The pith
Orbital-optimized density functional theory computes excited states by state-specific variational orbital optimization on the energy surface.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
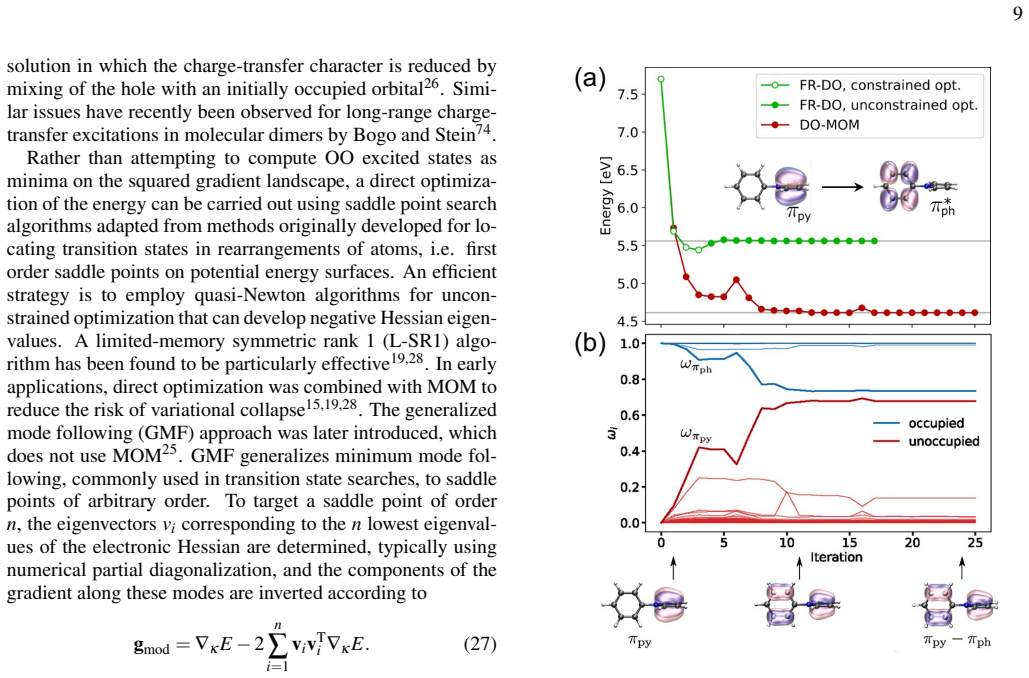
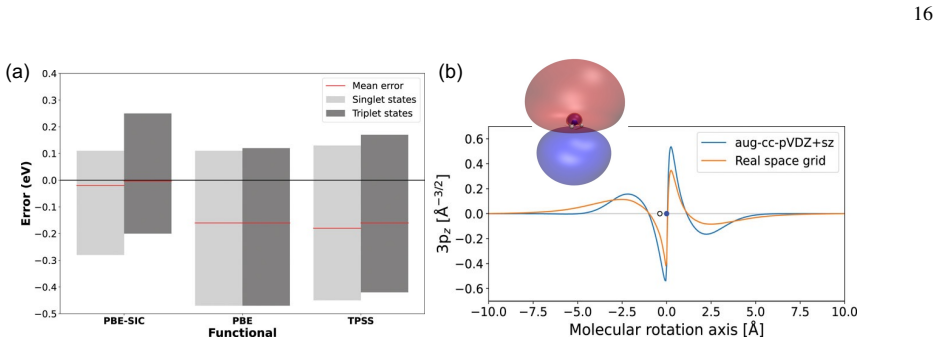
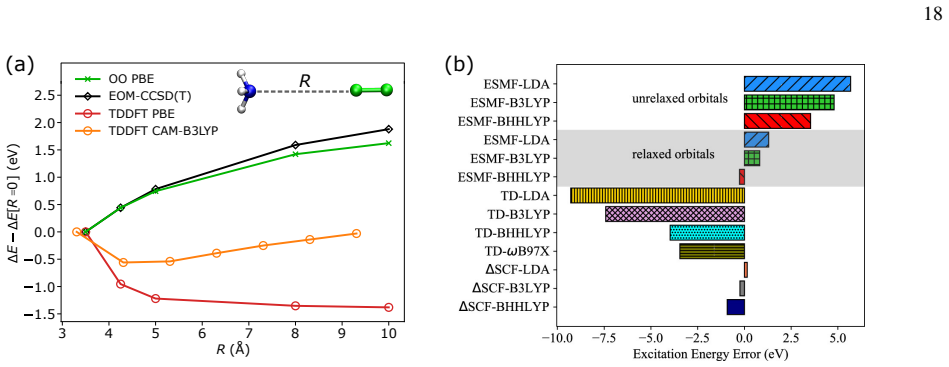
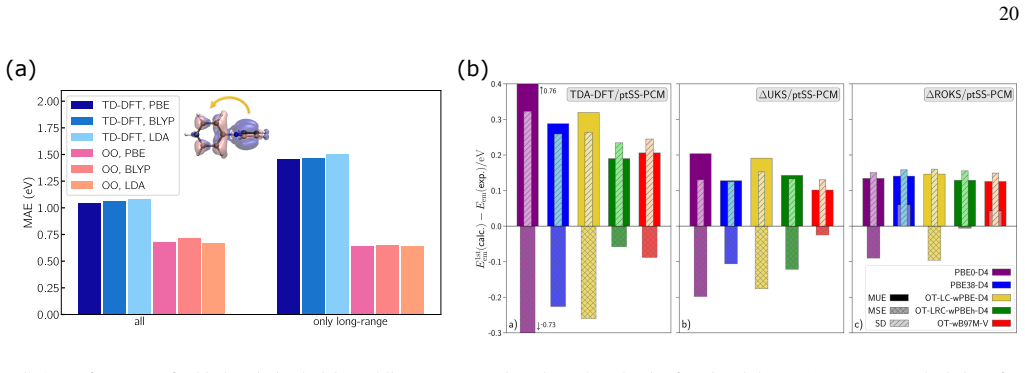
Orbital-optimized density functional calculations provide a time-independent, variational route to electronic excitations that complements TDDFT. As the orbitals are optimized in a state specific way, these methods can provide a balanced description of excited states with different character, thereby overcoming several limitations of practical implementations of TDDFT. Driven by recent developments in algorithms for obtaining excited states as saddle points on the electronic energy surface, OO methods have seen an increased interest in recent years, maturing into an active and rapidly developing area of research.
What carries the argument
State-specific orbital optimization treating each excited state as a saddle point on the electronic energy surface.
If this is right
- OO-DFT yields balanced descriptions for Rydberg, charge-transfer, and core excitations.
- Unified methods exist for treating open-shell singlet excited states.
- Transition properties and spectra can be obtained directly within the OO framework.
- Accuracy and applicability can be evaluated from recent molecular applications.
Where Pith is reading between the lines
- Further efficiency gains in saddle-point algorithms could extend OO-DFT to larger systems beyond current molecular examples.
- The variational character of OO-DFT may reduce reliance on response-theory instabilities in certain excitation regimes.
- Direct comparison of OO results against high-level wavefunction methods on the same test sets would sharpen assessments of functional performance.
Load-bearing premise
Recent algorithmic developments have matured OO methods into a reliable area whose accuracy can now be meaningfully assessed from published applications.
What would settle it
Systematic benchmarks across molecular Rydberg, charge-transfer, and core excitations that show OO-DFT fails to deliver more balanced accuracy than TDDFT for states of differing character.
Figures

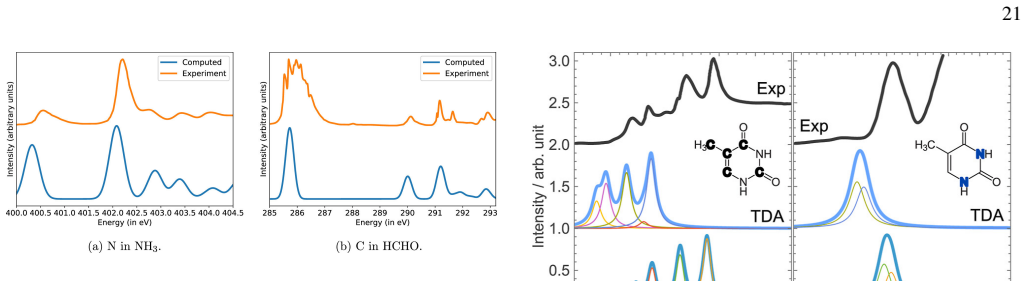
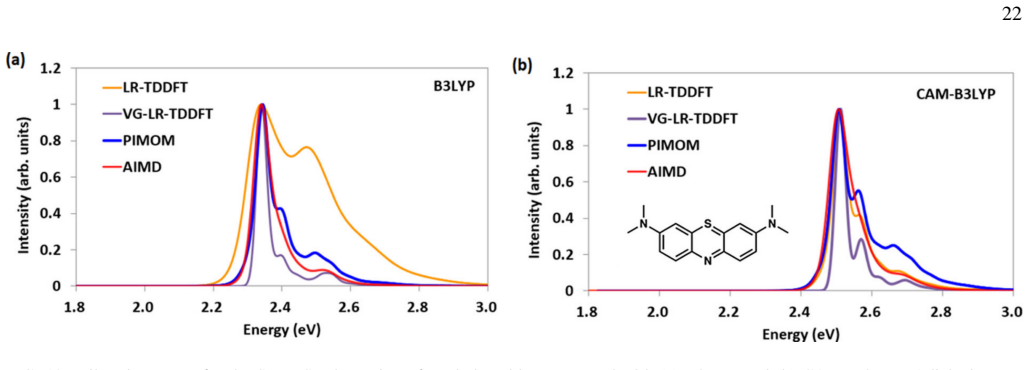
read the original abstract
Orbital-optimized (OO) density functional calculations provide a time-independent, variational route to electronic excitations that complements the presently widely used time-dependent density functional theory (TDDFT). As the orbitals are optimized in a state specific way, these methods can provide a balanced description of excited states with different character, thereby overcoming several limitations of practical implementations of TDDFT. Driven by recent developments in algorithms for obtained excited states as saddle points on the electronic energy surface, OO methods have seen an increased interest in recent years, maturing into an an active and rapidly developing area of research. Here, the theoretical foundations of the approach are clarified and an overview of the recent developments in methods for excited-state orbital optimization is provided. A unified overview of methods for treating open-shell singlet excited states and current approaches for computing transition properties and spectra is also provided. Finally, recent applications to molecular Rydberg, charge-transfer, and core excitations are reviewed, with the aim of assessing the present accuracy and range of applicability of OO density functional calculations.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript is a review article surveying orbital-optimized (OO) density functional theory methods for excited electronic states. It positions OO-DFT as a time-independent, variational complement to TDDFT that achieves balanced descriptions of excited states with differing characters via state-specific orbital optimization. The review clarifies theoretical foundations, covers recent algorithmic advances for locating excited states as saddle points, unifies treatments of open-shell singlet states and transition properties/spectra, and surveys applications to Rydberg, charge-transfer, and core excitations while assessing current accuracy and range of applicability.
Significance. If the synthesis is accurate and comprehensive, the review would be significant for computational chemistry by consolidating an active research area that addresses known TDDFT limitations for certain excitation types. The unified treatment of open-shell singlets, transition properties, and targeted application assessments could serve as a useful reference point for researchers seeking alternatives to linear-response methods.
minor comments (1)
- [Abstract] Abstract: the sentence 'maturing into an an active and rapidly developing area' contains a duplicated article ('an an'); this should be corrected to 'an active'.
Simulated Author's Rebuttal
We thank the referee for their positive assessment of the manuscript, the accurate summary of its scope, and the recommendation to accept.
Circularity Check
No significant circularity; review paper with no internal derivations or predictions
full rationale
This is a review article synthesizing existing literature on orbital-optimized DFT methods for excited states. It contains no new derivations, equations, or predictions that reduce to fitted inputs or self-citations by construction. The central claims are presented as overviews of published work rather than self-contained derivations, with no load-bearing steps that equate outputs to inputs within the paper. The reader's assessment of score 0.0 is confirmed by the absence of any mathematical chain or ansatz that could exhibit circularity.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Schmerwitz and Gianluca Levi , doi =
Elli Selenius and Alec Elías Sigurdarson and Yorick L.A. Schmerwitz and Gianluca Levi , doi =. Orbital-Optimized Versus Time-Dependent Density Functional Calculations of Intramolecular Charge Transfer Excited States , volume =. J. Chem. Theory Comput. , month =
-
[2]
PASC '24: Proceedings of the Platform for Advanced Scientific Computing Conference , pages =
Schmerwitz, Yorick Leonard Adrian and Oll. PASC '24: Proceedings of the Platform for Advanced Scientific Computing Conference , pages =. doi:2402.16601 , eprint =
-
[3]
Maria Hellgren and E. K.U. Gross , doi =. Discontinuities of the exchange-correlation kernel and charge-transfer excitations in time-dependent density-functional theory , volume =. Phys. Rev. A , month =
-
[4]
Stein , doi =
Nicola Bogo and Christopher J. Stein , doi =. Benchmarking DFT-based excited-state methods for intermolecular charge-transfer excitations , volume =. Phys. Chem. Chem. Phys. , month =
-
[5]
SciPost Phys
Electronic excitations of the charged nitrogen-vacancy center in diamond obtained using time-independent variational density functional calculations , author=. SciPost Phys. , volume=
-
[6]
Reference energies for intramolecular charge-transfer excitations , author=. J. Chem. Theory Comput. , volume=. 2021 , publisher=
2021
-
[7]
Solvent-Mediated Charge Transfer Dynamics of a Model Brown Carbon Aerosol Chromophore: Photophysics of 1-Phenylpyrrole Induced by Water Solvation , author=. J. Phys. Chem. A , year=
-
[8]
Charge-Transfer Excitations within Density Functional Theory: How Accurate Are the Most Recommended Approaches? , author=. J. Chem. Theory Comput. , volume=. 2022 , publisher=
2022
-
[9]
Journal of Photochemistry and Photobiology A: Chemistry , volume=
Performance of time-dependent density functional theory on twisted intramolecular charge transfer state of emerging visible light photoswitches , author=. Journal of Photochemistry and Photobiology A: Chemistry , volume=. 2019 , publisher=
2019
-
[10]
Benchmarking quantum mechanical methods for the description of charge-transfer states in -stacked nucleobases , author=. J. Chem. Theory Comput. , volume=. 2020 , publisher=
2020
-
[11]
Double hybrids and time-dependent density functional theory: An implementation and benchmark on charge transfer excited states , author=. J. Comput. Chem. , volume=. 2020 , publisher=
2020
-
[12]
Prediction of excited-state energies and singlet--triplet gaps of charge-transfer states using a restricted open-shell Kohn--Sham approach , author=. J. Chem. Theory Comput. , volume=. 2016 , publisher=
2016
-
[13]
Hait, Diptarka and Head-Gordon, Martin , doi =. J. Phys. Chem. Lett. , number =. 1912.05249 , issn =
arXiv 1912
-
[14]
Kohn and L
W. Kohn and L. J. Sham , title=. 1965 , journal=
1965
-
[15]
Predicting excitation energies of twisted intramolecular charge-transfer states with the time-dependent density functional theory: Comparison with experimental measurements in the gas phase and solvents ranging from hexanes to acetonitrile , author=. J. Chem. Theory Comput. , volume=. 2020 , publisher=
2020
-
[16]
Origin of the Failure of Density Functional Theories in Predicting Inverted Singlet--Triplet Gaps , author=. J. Phys. Chem. A , volume=. 2022 , publisher=
2022
-
[17]
Orbital optimized density functional theory for electronic excited states , author=. The J. Phys. Chem. Lett. , volume=. 2021 , publisher=
2021
-
[18]
Assessment of the SCF density functional theory approach for electronic excitations in organic dyes , author=. J. Chem. Phys. , volume=. 2011 , publisher=
2011
-
[19]
Kowalczyk, Tim and Tsuchimochi, Takashi and Chen, Po Ta and Top, Laken and. J. Chem. Phys. , number =. 2013 , pages=. doi:10.1063/1.4801790 , issn =
-
[20]
PCM-ROKS for the Description of Charge-Transfer States in Solution: Singlet--Triplet Gaps with Chemical Accuracy from Open-Shell Kohn--Sham Reaction-Field Calculations , author=. The J. Phys. Chem. Lett. , volume=. 2021 , publisher=
2021
-
[21]
The Journal of Physical Chemistry Letters , volume=
The best of both worlds: DFT describes multiresonance TADF emitters with wave-function accuracy at density-functional cost , author=. The Journal of Physical Chemistry Letters , volume=. 2025 , publisher=
2025
-
[22]
Physical Review A , volume=
Strategies for solving the excited-state self-consistent-field problem for highly excited and multiply ionized states , author=. Physical Review A , volume=. 2021 , publisher=
2021
-
[23]
Faraday Discussions , volume=
Variational calculations of excited states via direct optimization of the orbitals in DFT , author=. Faraday Discussions , volume=. 2020 , publisher=
2020
-
[24]
Variational density functional calculations of excited states via direct optimization , author=. J. Chem. Theory Comput. , volume=. 2020 , publisher=
2020
-
[25]
Ivanov, Aleksei V. and Levi, Gianluca and J. J. Chem. Theory Comput. , month =. doi:10.1021/acs.jctc.1c00157 , issn =
-
[26]
Variational Density Functional Calculations of Excited States: Conical Intersection and Avoided Crossing in Ethylene Bond Twisting , author=. The J. Phys. Chem. Lett. , volume=. 2022 , publisher=
2022
-
[27]
Rossi and Susi Lehtola and Arto Sakko and Martti J
Tuomas P. Rossi and Susi Lehtola and Arto Sakko and Martti J. Puska and Risto M. Nieminen , doi =. Nanoplasmonics simulations at the basis set limit through completeness-optimized, local numerical basis sets , volume =. J. Chem. Phys. , month =
-
[28]
A. H. Larsen and M. Vanin and J. J. Mortensen and K. S. Thygesen and K. W. Jacobsen , doi =. Localized atomic basis set in the projector augmented wave method , volume =. Phys. Rev. B, Condens. Matter , month =
-
[29]
and Gilbert, Andrew T.B
Barca, Giuseppe M.J. and Gilbert, Andrew T.B. and Gill, Peter M.W. , doi =. J. Chem. Theory Comput. , number =
-
[30]
Physical Review Letters , pages =
Thom, Alex J W and Head-Gordon, Martin , doi =. Physical Review Letters , pages =
-
[31]
Schmerwitz, Yorick L. A. and Levi, Gianluca and J. J. Chem. Theory Comput. , number =. doi:10.1021/acs.jctc.3c00178 , eprint =
-
[32]
Self-consistent field calculations of excited states using the maximum overlap method (MOM) , author=. J. Phys. Chem. A , volume=. 2008 , publisher=
2008
-
[33]
Failure of time-dependent density functional theory for long-range charge-transfer excited states: the zincbacteriochlorin- bacteriochlorin and bacteriochlorophyll- spheroidene complexes , author=. J. Am. Chem. Soc. , volume=. 2004 , publisher=
2004
-
[34]
and Head-Gordon, Martin , journal =
Dreuw, Andreas and Weisman, Jennifer L. and Head-Gordon, Martin , journal =
-
[35]
A SCF model for excited states within a polarisable embedding , author=. Mol. Phys. , pages=. 2022 , publisher=
2022
-
[36]
Mazzeo, Patrizia and Hashem, Shaima and Lipparini, Filippo and Cupellini, Lorenzo and Mennucci, Benedetta , doi =. J. Phys. Chem. Lett. , number =
-
[37]
Robust SCF calculations with direct energy functional minimization methods and STEP for molecules and materials , author=. J. Chem. Phys. , volume=. 2022 , publisher=
2022
-
[38]
The SCF method for non-adiabatic dynamics of systems in the liquid phase , author=. J. Chem. Phys. , volume=. 2022 , publisher=
2022
-
[39]
The photodissociation of solvated cyclopropanone and its hydrate explored via non-adiabatic molecular dynamics using SCF , author=. Phys. Chem. Chem. Phys. , volume=. 2022 , publisher=
2022
-
[40]
Simulation of Low-Lying Singlet and Triplet Excited States of Multiple-Resonance-Type Thermally Activated Delayed Fluorescence Emitters by Delta Self-Consistent Field ( SCF) Method , author=. J. Phys. Chem. A , volume=. 2021 , publisher=
2021
-
[41]
Relativistic orbital-optimized density functional theory for accurate core-level spectroscopy , author=. J. Phys. Chem. Lett. , volume=. 2022 , publisher=
2022
-
[42]
Journal of Chemical Theory and Computation , number =
Hait, Diptarka and Mart. Journal of Chemical Theory and Computation , number =. doi:10.1021/acs.jctc.3c01035 , issn =
-
[43]
Changes in polarization dictate necessary approximations for modeling electronic deexcitation intensity: Application to x-ray emission , author=. Phys. Rev. B , volume=. 2022 , publisher=
2022
-
[44]
Computing x-ray absorption spectra from linear-response particles atop optimized holes , author=. J. Chem. Phys. , volume=. 2022 , publisher=
2022
-
[45]
Jahn-Teller Distortion and Dissociation of CCl _4^
Ross, Andrew D and Hait, Diptarka and Scutelnic, Valeriu and Haugen, Eric A and Ridente, Enrico and Balkew, Mikias B and Neumark, Daniel M and Head-Gordon, Martin and Leone, Stephen R , journal=. Jahn-Teller Distortion and Dissociation of CCl _4^. 2022 , publisher =
2022
-
[46]
Rydberg energies using excited state density functional theory , author=. J. Chem. Phys. , volume=. 2008 , publisher=
2008
-
[47]
Density functional study of multiplicity-changing valence and Rydberg excitations of p-block elements: Delta self-consistent field, collinear spin-flip time-dependent density functional theory (DFT), and conventional time-dependent DFT , author=. J. Chem. Phys. , volume=. 2011 , publisher=
2011
-
[48]
Tautomerism in 5-methyltetrazole investigated by core-level photoelectron spectroscopy and SCF calculations , author=. Chem. Phys. Lett. , volume=. 2011 , publisher=
2011
-
[49]
Time-dependent density functional theory applied to ligand-field excitations and their circular dichroism in some transition metal complexes , author=. Chem. Phys. , volume=. 2011 , publisher=
2011
-
[50]
Assessing computationally efficient isomerization dynamics: SCF density-functional theory study of azobenzene molecular switching , author=. J. Chem. Phys. , volume=. 2011 , publisher=
2011
-
[51]
Role of electronic localization in the phosphorescence of iridium sensitizing dyes , author=. J. Chem. Phys. , volume=. 2012 , publisher=
2012
-
[52]
Applications of time-dependent and time-independent density functional theory to Rydberg transitions , author=. J. Phys. Chem. A , volume=. 2015 , publisher=
2015
-
[53]
Density functional theory based analysis of photoinduced electron transfer in a triazacryptand based K+ sensor , author=. J. Phys. Chem. A , volume=. 2015 , publisher=
2015
-
[54]
An illustration through applications to copper tetrachloride and plastocyanin , author=
A perspective on the relative merits of time-dependent and time-independent density functional theory in studies of the electron spectra due to transition metal complexes. An illustration through applications to copper tetrachloride and plastocyanin , author=. Int. J. Quantum Chem. , volume=. 2014 , publisher=
2014
-
[55]
Applications of time dependent and time independent density functional theory to the first to * transition in cyanine dyes , author=. J. Chem. Theory Comput. , volume=. 2014 , publisher=
2014
-
[56]
Dyes Pigm
Intramolecular charge transfer tuning of azo dyes: spectroscopic characteristic and third-order nonlinear optical properties , author=. Dyes Pigm. , volume=. 2018 , publisher=
2018
-
[57]
Non-adiabatic molecular dynamics with SCF excited states , author=. J. Phys. Condens. Matter , volume=. 2018 , publisher=
2018
-
[58]
An efficient first principles method for molecular pump-probe NEXAFS spectra: Application to thymine and azobenzene , author=. J. Chem. Phys. , volume=. 2018 , publisher=
2018
-
[59]
Self-consistent-field calculations of core excited states , author=. J. Chem. Phys. , volume=. 2009 , publisher=
2009
-
[60]
Solution structure and ultrafast vibrational relaxation of the PtPOP complex revealed by SCF-QM/MM direct dynamics simulations , author=. J. Phys. Chem. C , volume=. 2018 , publisher=
2018
-
[61]
Theoretical insights into the phosphorescent process of a series of 2-(2-trifluoromethyl) pyrimidine-pyridine based heteroleptic iridium (III) compounds: The influence of the ancillary ligand , author=. Comput. Theor. Chem. , volume=. 2017 , publisher=
2017
-
[62]
Excited states of light-harvesting systems based on fullerene/graphene oxide and porphyrin/smaragdyrin , author=. J. Phys. Chem. C , volume=. 2017 , publisher=
2017
-
[63]
Reliable transition properties from excited-state mean-field calculations , author=. J. Chem. Phys. , volume=. 2021 , publisher=
2021
-
[64]
SCF with Subsystem Density Embedding for Efficient Nonadiabatic Molecular Dynamics in Condensed-Phase Systems , author=. J. Chem. Theory Comput. , volume=
-
[65]
State-targeted energy projection: A simple and robust approach to orbital relaxation of non-Aufbau self-consistent field solutions , author=. J. Chem. Theory Comput. , volume=. 2020 , publisher=
2020
-
[66]
On the molecular mechanism of non-radiative decay of nitrobenzene and the unforeseen challenges this simple molecule holds for electronic structure theory , author=. Phys. Chem. Chem. Phys. , volume=. 2014 , publisher=
2014
-
[67]
Trajectory Surface Hopping Nonadiabatic Molecular Dynamics with Kohn--Sham SCF for Condensed-Phase Systems , author=. J. Chem. Theory Comput. , volume=. 2020 , publisher=
2020
-
[68]
Charge transfer excited state energies by perturbative delta self consistent field method , author=. J. Chem. Phys. , volume=. 2012 , publisher=
2012
-
[69]
Zn (II)-Porphyrin--Squaraine Dyads as Potential Components for Dye-Sensitized Solar Cells: A Quantum Chemical Study of Optical and Charge Transport Properties , author=. J. Phys. Chem. C , volume=. 2020 , publisher=
2020
-
[70]
Predicting core level photoelectron spectra of amino acids using density functional theory , author=. J. Phys. Chem. Lett. , volume=. 2020 , publisher=
2020
-
[71]
Excited state orbital optimization via minimizing the square of the gradient: General approach and application to singly and doubly excited states via density functional theory , author=. J. Chem. Theory Comput. , volume=. 2020 , publisher=
2020
-
[72]
Simple models for difficult electronic excitations , author=. J. Chem. Theory Comput. , volume=. 2018 , publisher=
2018
-
[73]
Low energy excitations in NiO based on a direct -SCF approach , author=. J. Phys. Condens. Matter , volume=. 2018 , publisher=
2018
-
[74]
On the origin of the inverted singlet--triplet gap of the 5th generation light-emitting molecules , author=. Phys. Chem. Chem. Phys. , volume=. 2022 , publisher=
2022
-
[75]
The Journal of Physical Chemistry A , month =
Toffoli, Daniele and Quarin, Matteo and Fronzoni, Giovanna and Stener, Mauro , doi =. The Journal of Physical Chemistry A , month =
-
[76]
Journal of Chemical Theory and Computation , month =
Mali. Journal of Chemical Theory and Computation , month =. doi:10.1021/ACS.JCTC.1C01046/ASSET/IMAGES/MEDIUM/CT1C01046_M066.GIF , issn =
work page doi:10.1021/acs.jctc.1c01046/asset/images/medium/ct1c01046_m066.gif
-
[77]
Recent advances in organic light-emitting diodes: toward smart lighting and displays , author=. Mater. Chem. Front. , volume=. 2020 , publisher=
2020
-
[78]
Molecular motion in aggregates: manipulating TICT for boosting photothermal theranostics , author=. J. Am. Chem. Soc. , volume=. 2019 , publisher=
2019
-
[79]
All-organic thermally activated delayed fluorescence materials for organic light-emitting diodes , author=. Nat. Rev. Mater. , volume=. 2018 , publisher=
2018
-
[80]
Green Chem
Recent advances in eco-friendly and cost-effective materials towards sustainable dye-sensitized solar cells , author=. Green Chem. , volume=. 2020 , publisher=
2020
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.