PairSAE: Mechanistic Interpretability from Pair Representations in Protein Co-Folding
Pith reviewed 2026-06-29 01:21 UTC · model grok-4.3
The pith
PairSAE produces interpretable features from pair representations in protein co-folding models by summarizing tensors with N-mode SVD before sparse autoencoding.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
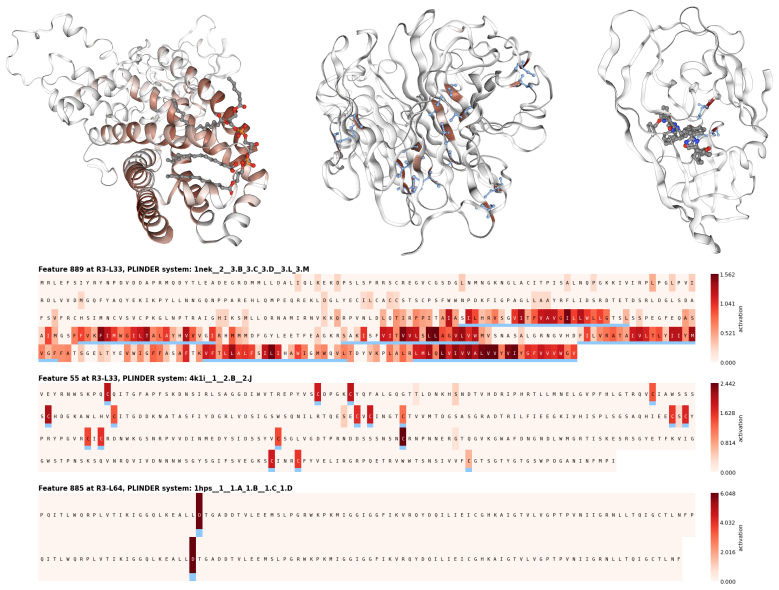
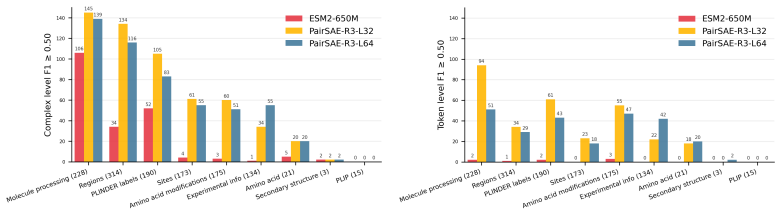
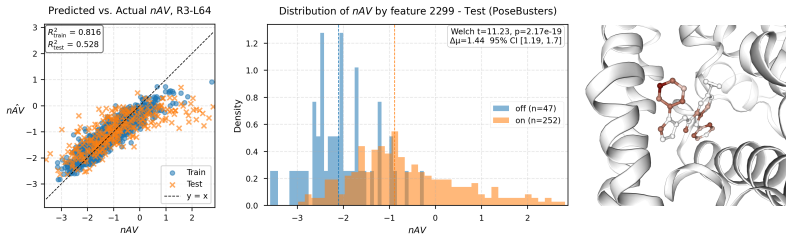
PairSAE summarizes pairwise tensors via an N-mode SVD into token-wise interaction roles, then uses a sparse autoencoder to learn a shared set of token-level features that decode into both sequence and pair representations. Evaluated on Boltz-2 activations for PLINDER protein-ligand complexes, PairSAE yields interpretable features that align with UniProt annotations and predict Boltz-2 affinity values.
What carries the argument
N-mode SVD summarization of pairwise tensors into token-wise interaction roles, followed by a shared sparse autoencoder that reconstructs both sequence and pair data.
If this is right
- Features align with existing UniProt annotations on protein-ligand complexes.
- The same features predict numerical affinity values from the model.
- The method avoids the quadratic feature blow-up and loss of joint concepts that occur with naive application of SAEs to pair tensors.
- It supplies a route from foundation-model activations to human-readable structural biology concepts.
Where Pith is reading between the lines
- The same SVD-plus-shared-SAE pattern could be tested on other pair-based architectures outside structural biology, such as graph transformers or vision-language models.
- The recovered features might be used to locate and correct specific failure modes in the original model on particular interaction types.
- If the features prove stable across different training runs of the base model, they could serve as a diagnostic for whether the foundation model has internalized particular biological rules.
Load-bearing premise
That reducing pairwise tensors to token-wise roles via N-mode SVD still lets a sparse autoencoder recover distributed concepts that are jointly encoded across sequence and pair representations.
What would settle it
If the extracted features show no statistical alignment with UniProt annotations or no above-chance correlation with Boltz-2 affinity values on held-out complexes, the claim that PairSAE links the latent space to interpretable structural concepts would not hold.
Figures
read the original abstract
Foundation models for structural biology have achieved remarkable performance in predicting biomolecular structure and show promise for the design of proteins and small molecules. Yet understanding which internal features drive their outputs remains challenging. Standard sparse autoencoders (SAEs), effective on transformer-style sequence embeddings, do not transfer cleanly to pairformer-like architectures: naively operating on pairwise representations yields a quadratic blow-up of features and obscures concepts distributed jointly across sequence and pair representations. We introduce PairSAE, which summarizes pairwise tensors via an N-mode SVD into token-wise interaction roles, then uses a sparse autoencoder to learn a shared set of token-level features that decode into both sequence and pair representations. Evaluated on Boltz-2 activations for PLINDER protein-ligand complexes, PairSAE yields interpretable features that align with UniProt annotations and predict Boltz-2 affinity values. These results indicate that PairSAE links the latent space of foundation models for structural biology to interpretable structural concepts, clarifying what the model "knows" while avoiding pairformer-induced pitfalls that limit conventional SAEs.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces PairSAE to enable sparse autoencoder-based interpretability on pair representations from pairformer-style models in structural biology. Pairwise tensors are reduced via N-mode SVD to token-wise interaction roles; an SAE then learns a shared dictionary of token-level features that are decoded back into both sequence and pair representations. On Boltz-2 activations from PLINDER protein-ligand complexes, the resulting features are reported to align with UniProt annotations and to predict affinity values, thereby linking model latents to interpretable structural concepts while sidestepping quadratic feature blow-up.
Significance. If the SVD reduction demonstrably preserves distributed joint sequence-pair concepts and the reported alignments hold under quantitative controls, the work would supply a practical route for mechanistic interpretability in co-folding foundation models, a domain where standard SAEs have been limited by representation geometry.
major comments (2)
- [Abstract] Abstract and implied Methods: the central claim that PairSAE 'avoids pairformer-induced pitfalls' and recovers features that 'decode into both sequence and pair representations' without loss of distributed concepts rests on the untested assumption that N-mode SVD truncation to token-wise roles preserves non-factorizable pairwise patterns (e.g., cooperative motifs). No reconstruction fidelity metrics, ablation on SVD rank, or comparison of pair-reconstruction error before versus after the reduction are supplied to substantiate this preservation.
- [Abstract] Abstract: the evaluation claims that features 'align with UniProt annotations and predict Boltz-2 affinity values' but supplies neither dataset sizes, train/test splits, quantitative metrics (R², AUROC, error bars), nor controls for spurious correlation; without these the load-bearing assertion that the features are mechanistically meaningful cannot be assessed.
minor comments (1)
- [Abstract] Acronyms Boltz-2 and PLINDER are used without expansion on first use.
Simulated Author's Rebuttal
We thank the referee for the constructive feedback. The two major comments identify gaps in quantitative validation of the N-mode SVD step and in the reporting of evaluation details. We will revise the manuscript to incorporate the requested analyses and metrics. Point-by-point responses are below.
read point-by-point responses
-
Referee: [Abstract] Abstract and implied Methods: the central claim that PairSAE 'avoids pairformer-induced pitfalls' and recovers features that 'decode into both sequence and pair representations' without loss of distributed concepts rests on the untested assumption that N-mode SVD truncation to token-wise roles preserves non-factorizable pairwise patterns (e.g., cooperative motifs). No reconstruction fidelity metrics, ablation on SVD rank, or comparison of pair-reconstruction error before versus after the reduction are supplied to substantiate this preservation.
Authors: We agree that explicit validation of the SVD reduction is required to support the claim that distributed pairwise concepts are preserved. The N-mode SVD is motivated as a low-rank factorization that isolates token-wise interaction roles while retaining the dominant joint sequence-pair structure, but this is an assumption that needs empirical backing. In the revised manuscript we will add (i) pair-tensor reconstruction error (Frobenius norm) before versus after truncation, (ii) an ablation over SVD rank showing the trade-off between compression and fidelity, and (iii) qualitative examples of preserved cooperative motifs. These additions will directly address the concern. revision: yes
-
Referee: [Abstract] Abstract: the evaluation claims that features 'align with UniProt annotations and predict Boltz-2 affinity values' but supplies neither dataset sizes, train/test splits, quantitative metrics (R², AUROC, error bars), nor controls for spurious correlation; without these the load-bearing assertion that the features are mechanistically meaningful cannot be assessed.
Authors: We acknowledge that the current abstract and main text omit the requested quantitative details. The evaluations were performed on the PLINDER protein-ligand complexes using Boltz-2 activations, but the manuscript does not report dataset cardinality, splits, or statistical controls. In revision we will explicitly state the number of complexes, the train/validation/test partitioning, the precise metrics (R² for affinity regression, AUROC for UniProt annotation alignment) with error bars across seeds, and negative controls (random features and label-shuffled baselines) to demonstrate that alignments exceed spurious correlation. These changes will make the mechanistic claims assessable. revision: yes
Circularity Check
No circularity: method proposal contains no derivations or self-referential reductions
full rationale
The provided abstract and description introduce PairSAE as a procedural pipeline (N-mode SVD summarization followed by SAE training on token-wise roles) without any equations, fitted parameters renamed as predictions, self-citations, or uniqueness theorems. No load-bearing step reduces a claimed result to its own inputs by construction; the work is a self-contained methodological proposal whose validity rests on empirical evaluation rather than internal definitional closure.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
Boltz-1 democratizing biomolecular interaction modeling.BioRxiv, pages 2024–11,
Jeremy Wohlwend, Gabriele Corso, Saro Passaro, Noah Getz, Mateo Reveiz, Ken Leidal, Wojtek Swiderski, Liam Atkinson, Tally Portnoi, Itamar Chinn, et al. Boltz-1 democratizing biomolecular interaction modeling.BioRxiv, pages 2024–11,
2024
-
[2]
Boltz-2: Towards accurate and efficient binding affinity prediction.BioRxiv, pages 2025–06,
Saro Passaro, Gabriele Corso, Jeremy Wohlwend, Mateo Reveiz, Stephan Thaler, Vignesh Ram Somnath, Noah Getz, Tally Portnoi, Julien Roy, Hannes Stark, et al. Boltz-2: Towards accurate and efficient binding affinity prediction.BioRxiv, pages 2025–06,
2025
-
[3]
Nelson Elhage, Tristan Hume, Catherine Olsson, Nicholas Schiefer, Tom Henighan, Shauna Kravec, Zac Hatfield-Dodds, Robert Lasenby, Dawn Drain, Carol Chen, et al. Toy models of superposition. arXiv preprint arXiv:2209.10652,
work page internal anchor Pith review Pith/arXiv arXiv
-
[4]
Polysemanticity and capacity in neural networks.arXiv preprint arXiv:2210.01892,
Adam Scherlis, Kshitij Sachan, Adam S Jermyn, Joe Benton, and Buck Shlegeris. Polysemanticity and capacity in neural networks.arXiv preprint arXiv:2210.01892,
-
[5]
Sparse autoencoder.CS294A Lecture notes, 72(2011):1–19,
Andrew Ng et al. Sparse autoencoder.CS294A Lecture notes, 72(2011):1–19,
2011
-
[6]
Burke, Tristan Hume, Shan Carter, Tom Henighan, and Christopher Olah
Trenton Bricken, Adly Templeton, Joshua Batson, Brian Chen, Adam Jermyn, Tom Conerly, Nick Turner, Cem Anil, Carson Denison, Amanda Askell, Robert Lasenby, Yifan Wu, Shauna Kravec, Nicholas Schiefer, Tim Maxwell, Nicholas Joseph, Zac Hatfield-Dodds, Alex Tamkin, Karina Nguyen, Brayden McLean, Josiah E. Burke, Tristan Hume, Shan Carter, Tom Henighan, and C...
2023
-
[7]
Elana Simon and James Zou
Transformer Circuits Thread. Elana Simon and James Zou. Interplm: Discovering interpretable features in protein language models via sparse autoencoders.bioRxiv, pages 2024–11,
2024
-
[8]
Edith Natalia Villegas Garcia and Alessio Ansuini
URL https://transformer-circuits.pub/2024/scaling-monosemanticity/ index.html. Edith Natalia Villegas Garcia and Alessio Ansuini. Interpreting and steering protein language models through sparse autoencoders.arXiv preprint arXiv:2502.09135,
-
[9]
Towards interpretable protein structure prediction with sparse autoencoders
6 Nithin Parsan, David J Yang, and John Jingxuan Yang. Towards interpretable protein structure prediction with sparse autoencoders. InLearning Meaningful Representations of Life (LMRL) Workshop at ICLR 2025,
2025
-
[10]
Genome modeling and design across all domains of life with evo 2.BioRxiv, pages 2025–02,
Garyk Brixi, Matthew G Durrant, Jerome Ku, Michael Poli, Greg Brockman, Daniel Chang, Gabriel A Gonzalez, Samuel H King, David B Li, Aditi T Merchant, et al. Genome modeling and design across all domains of life with evo 2.BioRxiv, pages 2025–02,
2025
-
[11]
Bart Bussmann, Patrick Leask, and Neel Nanda
Bart Bussmann, Patrick Leask, and Neel Nanda. Batchtopk sparse autoencoders.arXiv preprint arXiv:2412.06410,
-
[12]
Plinder: The protein-ligand interactions dataset and evaluation resource.bioRxiv, pages 2024–07,
Janani Durairaj, Yusuf Adeshina, Zhonglin Cao, Xuejin Zhang, Vladas Oleinikovas, Thomas Duignan, Zachary McClure, Xavier Robin, Gabriel Studer, Daniel Kovtun, et al. Plinder: The protein-ligand interactions dataset and evaluation resource.bioRxiv, pages 2024–07,
2024
-
[13]
Uniprot: the universal protein knowledgebase in 2025.Nucleic acids research, 53(D1): D609–D617,
UniProt. Uniprot: the universal protein knowledgebase in 2025.Nucleic acids research, 53(D1): D609–D617,
2025
-
[14]
Adam: A Method for Stochastic Optimization
Diederik P Kingma and Jimmy Ba. Adam: A method for stochastic optimization.arXiv preprint arXiv:1412.6980,
work page internal anchor Pith review Pith/arXiv arXiv
-
[15]
Following De Lathauwer et al
8 AN-mode Singular Value Decomposition The mode-k unfolding of a tensor T ∈R n1×n2×···×nN is obtained by flattening all but the kth dimension into a matrix T (k) ∈R nk×Q i̸=k ni. Following De Lathauwer et al. [2000a], every tensor admits the higher-order singular value decomposition T=C × 1 U(1) ×2 U(2) · · · ×N U(N) , where C ∈R n1×n2×···×nN is a core te...
2002
-
[16]
We truncate to the first r= 64 columns, and if Ntok < r we fill the remaining entries with zeroes
We compute the N−mode SVD by simply flattening Z to its mode-1 and mode-2 unfolding, and obtain the SVD of these matrices using numpy.linalg.svd. We truncate to the first r= 64 columns, and if Ntok < r we fill the remaining entries with zeroes. After concatenating sequence embeddings s and the SVD-derived embedding m into a 512-dimensional vector, we perf...
2014
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.