An Additive Reference Correction Scheme for the Transcorrelated Method
Pith reviewed 2026-07-01 03:05 UTC · model grok-4.3
The pith
Replacing the reference energy in xTC calculations with a larger-basis value improves atomization energies in double-ζ bases while keeping the small-basis correlation energy.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
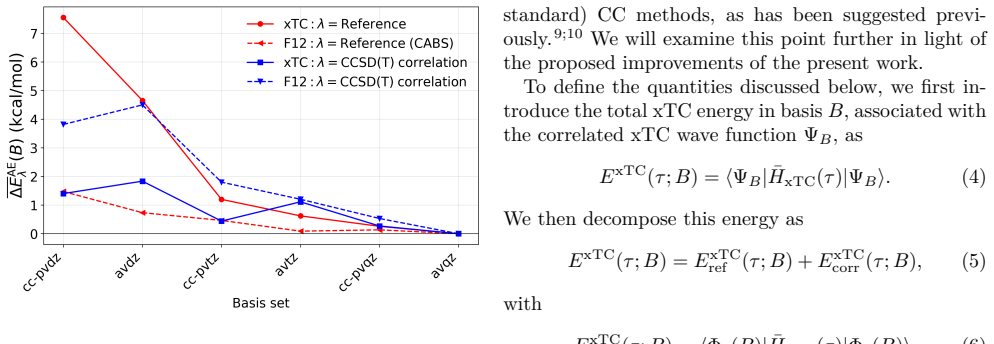
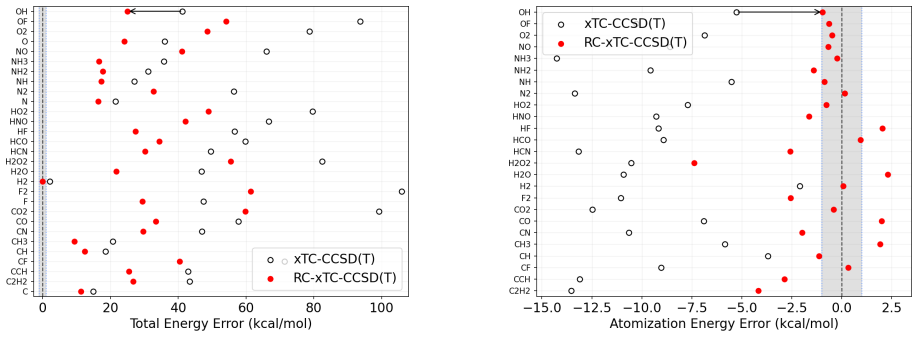
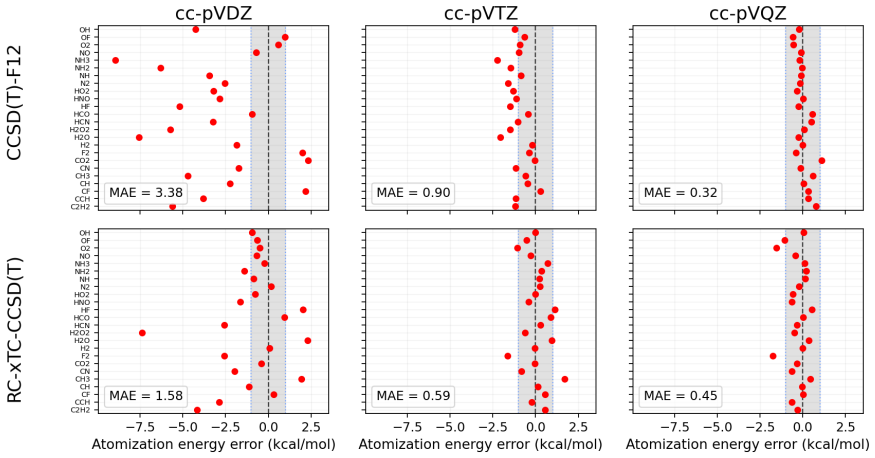
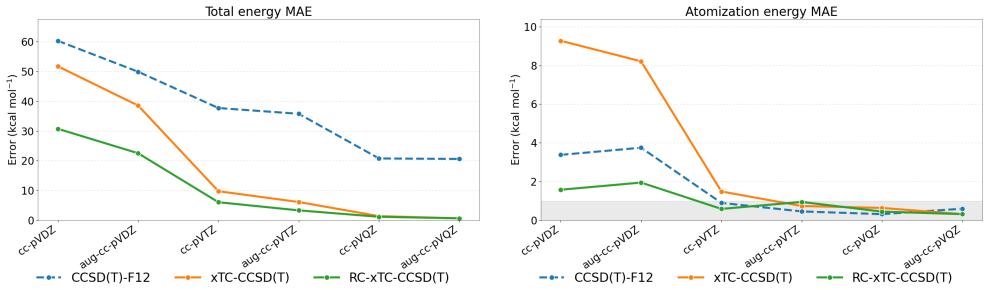
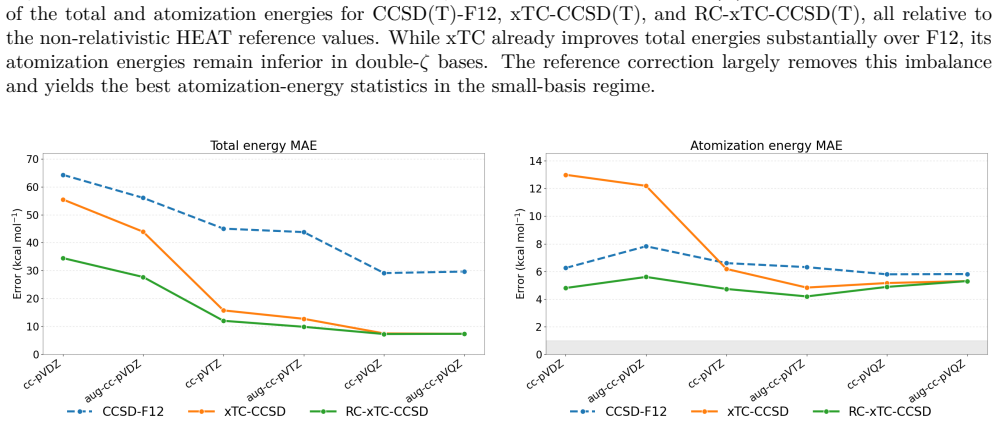
The central claim is that for xTC atomization energies the dominant error in double-ζ bases originates from the reference contribution rather than from the correlation energy. In the reference-corrected scheme the small-basis correlation energy is retained while the TC reference energy is replaced by its larger-basis counterpart. Benchmark calculations for the HEAT set with the Dunning family show that this RC-xTC scheme substantially improves both total and atomization energies relative to standard xTC in double-ζ bases. At the CCSD(T) level RC-xTC yields better atomization energies than CCSD(T)-F12a in the double-ζ regime while preserving the favorable total-energy accuracy of xTC; at the
What carries the argument
The RC-xTC scheme, an additive correction that substitutes the small-basis TC reference energy with its larger-basis value while retaining the small-basis correlation energy.
If this is right
- RC-xTC improves both total and atomization energies relative to standard xTC in double-ζ bases on the HEAT set.
- At the CCSD(T) level, RC-xTC produces better atomization energies than CCSD(T)-F12a in the double-ζ regime while retaining xTC total-energy accuracy.
- At the CCSD level, RC-xTC improves atomization energies relative to F12a across the full basis-set sequence.
- As the basis set enlarges, RC-xTC converges to standard xTC by construction.
Where Pith is reading between the lines
- The same reference-swap idea could be tested on other energy differences such as reaction barriers where reference contributions might similarly dominate.
- Applying the correction to even smaller bases such as single-ζ could check whether the reference-error dominance persists and how far the scheme remains useful.
- The construction suggests that separating reference and correlation treatments can be an efficient route for transcorrelated methods when basis-set limitations are the main bottleneck.
Load-bearing premise
The dominant error for xTC atomization energies in double-ζ bases originates from the reference contribution rather than from the correlation energy.
What would settle it
A decomposition of the xTC atomization energy error in double-ζ bases that shows the correlation-energy contribution exceeds the reference-energy contribution would falsify the premise for the correction.
Figures

read the original abstract
We introduce an additive reference correction for the transcorrelated (TC) method and its three-body mean-field approximation (xTC), to improve energy differences computed in small orbital basis sets. The correction is motivated by the observation that, for xTC atomization energies, the dominant error in double-{\zeta} bases originates from the reference contribution rather than from the correlation energy. In the proposed reference-corrected scheme (RC-xTC), the small-basis correlation energy is retained, while the corresponding TC reference energy is replaced by its value from a larger basis. Benchmark calculations for the non-relativistic HEAT set with the Dunning basis-set family show that RC-xTC substantially improves both total and atomization energies relative to standard xTC in double-{\zeta} bases. At the CCSD(T) level, RC-xTC yields better atomization energies than CCSD(T)-F12a in the double-{\zeta} regime, while preserving the favorable total-energy accuracy of xTC. At the CCSD level, RC-xTC improves atomization energies relative to F12a throughout the full basis-set sequence. As the basis set is enlarged, xTC and RC-xTC become progressively identical, as expected from the construction of the correction.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces an additive reference correction scheme (RC-xTC) for the transcorrelated (TC) method and its three-body mean-field approximation (xTC). The scheme retains the small-basis correlation energy but replaces the TC reference energy with its value computed in a larger basis set. This is motivated by the observation that, for xTC atomization energies, the dominant error in double-ζ bases originates from the reference rather than the correlation contribution. Benchmark calculations on the non-relativistic HEAT set using the Dunning basis-set family demonstrate that RC-xTC substantially improves both total and atomization energies relative to standard xTC in double-ζ bases. At the CCSD(T) level, RC-xTC yields better atomization energies than CCSD(T)-F12a in the double-ζ regime while preserving the favorable total-energy accuracy of xTC. At the CCSD level, improvements over F12a are seen across the basis-set sequence.
Significance. If the central results hold, the RC-xTC scheme offers a computationally efficient way to enhance the accuracy of transcorrelated calculations in small orbital bases without modifying the correlation treatment. The direct comparisons to explicitly correlated F12a methods on the HEAT set provide useful benchmarks for the community. The approach becomes equivalent to standard xTC as the basis set is enlarged, consistent with its construction. The paper provides concrete numerical evidence of improvements in both total and atomization energies.
major comments (1)
- [Introduction] Introduction (and results section): The central motivation for retaining the small-basis correlation energy unchanged rests on the claim that reference errors dominate over correlation errors for xTC atomization energies in double-ζ bases. However, no explicit decomposition of basis-set errors into reference and correlation components is provided for the HEAT set molecules, so it remains possible that the reported net improvements arise from a different balance of errors.
minor comments (1)
- [Abstract] Abstract: The statement of the motivating observation does not include a cross-reference to the specific results, table, or figure where this dominance is demonstrated.
Simulated Author's Rebuttal
We thank the referee for their careful reading of the manuscript and for the constructive feedback. We address the single major comment below.
read point-by-point responses
-
Referee: [Introduction] Introduction (and results section): The central motivation for retaining the small-basis correlation energy unchanged rests on the claim that reference errors dominate over correlation errors for xTC atomization energies in double-ζ bases. However, no explicit decomposition of basis-set errors into reference and correlation components is provided for the HEAT set molecules, so it remains possible that the reported net improvements arise from a different balance of errors.
Authors: We agree that an explicit decomposition of the basis-set errors would strengthen the motivation for the RC-xTC scheme. In the revised manuscript we will add this decomposition (as a table in the results section) for the HEAT set at the double-ζ level, separating the reference and correlation contributions to both total and atomization energies. This will directly demonstrate that the reference error dominates for xTC atomization energies and thereby justify retaining the small-basis correlation energy. revision: yes
Circularity Check
No significant circularity; scheme is an explicit additive definition with independent benchmarks
full rationale
The RC-xTC method is defined directly by replacing the small-basis TC reference energy with its large-basis counterpart while retaining the small-basis correlation energy; this is a constructional choice, not a derivation that reduces to its own inputs by equation. The paper reports numerical improvements on the external HEAT benchmark set against CCSD(T)-F12a and other methods, providing falsifiable evidence outside the definition itself. No self-citations, fitted parameters renamed as predictions, or uniqueness theorems are invoked in the abstract or described construction. The motivating observation about error dominance is stated but does not create a self-referential loop in the reported results.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption For xTC atomization energies the dominant error in double-ζ bases originates from the reference contribution rather than from the correlation energy.
Reference graph
Works this paper leans on
-
[1]
S. F. Boys and N. C. Handy. A calculation for the energies and wavefunctions for states of neon with full electronic correlation accuracy.Proceedings of the Royal Society of London. Series A, Mathemat- ical and Physical Sciences, 310:63–78, 1969. doi: 10.1098/rspa.1969.0062
-
[2]
N. C. Handy. Energies and expectation values for be by the transcorrelated method.The Journal of Chemical Physics, 51(8):3205–3212, 1969. doi: 10.1063/1.1672496
-
[3]
Hongjun Luo and Ali Alavi. Combining the transcorrelated method with full configuration in- teraction quantum monte carlo: Application to the homogeneous electron gas.Journal of Chemical Theory and Computation, 14(3):1403–1411, 2018. doi: 10.1021/acs.jctc.7b01257
-
[4]
Philip Haupt, Seyed Mohammadreza Hosseini, Pablo López Ríos, Werner Dobrautz, Aron J
J. Philip Haupt, Seyed Mohammadreza Hosseini, Pablo López Ríos, Werner Dobrautz, Aron J. Cohen, and Ali Alavi. Optimizing jastrow fac- tors for the transcorrelated method.The Journal 6 of Chemical Physics, 158(22):224105, 2023. doi: 10.1063/5.0147877
-
[5]
Evelin Martine Corvid Christlmaier, Thomas Schraivogel, Pablo López Ríos, Ali Alavi, and Daniel Kats. xtc: An efficient treatment of three- body interactions in transcorrelated methods.The Journal of Chemical Physics, 159(1):014113, 2023. doi: 10.1063/5.0154445
-
[6]
Philip Haupt, Evelin Martine Corvid Christlmaier, Daniel Kats, and Ali Alavi
Maria-Andreea Filip, Pablo López Ríos, J. Philip Haupt, Evelin Martine Corvid Christlmaier, Daniel Kats, and Ali Alavi. Transcorrelated methods applied to second row elements.The Journal of Chemical Physics, 162(6):064110, 2025. doi: 10.1063/5.0246422
-
[7]
Transcorre- lated density matrix renormalization group.The Journal of Chemical Physics, 153(16):164115, 10
Alberto Baiardi and Markus Reiher. Transcorre- lated density matrix renormalization group.The Journal of Chemical Physics, 153(16):164115, 10
- [8]
-
[9]
Ke Liao, Huanchen Zhai, Evelin Martine Corvid Christlmaier, Thomas Schraivogel, Pablo López Ríos, Daniel Kats, and Ali Alavi. Density matrix renormalization group for transcorre- lated hamiltonians: Ground and excited states in molecules.Journal of Chemical Theory and Computation, 19(6):1734–1743, 2023. doi: 10.1021/acs.jctc.2c01207
-
[10]
Cohen, Ali Alavi, and Daniel Kats
Thomas Schraivogel, Aron J. Cohen, Ali Alavi, and Daniel Kats. Transcorrelated coupled cluster meth- ods.The Journal of Chemical Physics, 155(19): 191101, 2021. doi: 10.1063/5.0072495
-
[11]
Transcorrelated coupled cluster methods
Thomas Schraivogel, Evelin Martine Corvid Christlmaier, Pablo López Ríos, Ali Alavi, and Daniel Kats. Transcorrelated coupled cluster methods. ii. molecular systems.The Journal of Chemical Physics, 158(21):214106, 2023. doi: 10.1063/5.0151412
-
[12]
Or- bital optimisation in xTC transcorrelated meth- ods.Faraday Discussions, 254(0):382, 2024
Daniel Kats, Evelin Martine Corvid Christl- maier, Thomas Schraivogel, and Ali Alavi. Or- bital optimisation in xTC transcorrelated meth- ods.Faraday Discussions, 254(0):382, 2024. doi: 10.1039/D4FD00036F
-
[13]
Tc++: First-principles calculation code for solids using the transcorrelated method
Masayuki Ochi. Tc++: First-principles calculation code for solids using the transcorrelated method. Computer Physics Communications, 287:108687,
-
[14]
doi: 10.1016/j.cpc.2023.108687
-
[15]
Shinji Tsuneyuki. Transcorrelated method: An- other possible way towards electronic structure calculation of solids.Progress of Theoretical Physics Supplement, 176:134–142, 2008. doi: 10.1143/PTPS.176.134
-
[16]
Kristoffer Simula, Johannes Hauskrecht, Evelin Martine Corvid Christlmaier, Pablo Lopez-Rios, Daniel Kats, Denis Usvyat, and Ali Alavi. Transcorrelated wave-function framework for solids: An application to bulk and defected sili- con.Phys. Rev. B, 113:195125, May 2026. doi: 10.1103/d65l-5865
-
[17]
Transcorrelated ap- proach to the thermodynamic limit.Physical Re- view B, 113(16):165146, 2026
Hongjun Luo and Ali Alavi. Transcorrelated ap- proach to the thermodynamic limit.Physical Re- view B, 113(16):165146, 2026. doi: 10.1103/jwz7- f23z
-
[18]
Nicholas Lee and Alex J. W. Thom. Studies on the transcorrelated method.Journal of Chemical Theory and Computation, 19(17):5743–5759, 2023. doi: 10.1021/acs.jctc.3c00046
-
[19]
Abdallah Ammar, Anthony Scemama, and Em- manuel Giner. Biorthonormal orbital optimiza- tion with a cheap core-electron-free three-body correlation factor for quantum monte carlo and transcorrelation.Journal of Chemical Theory and Computation, 19(15):4883–4896, 2023. doi: 10.1021/acs.jctc.3c00257
-
[20]
Compactifica- tionofdeterminantexpansionsviatranscorrelation
Abdallah Ammar, Anthony Scemama, Pierre- FrançoisLoos, andEmmanuelGiner. Compactifica- tionofdeterminantexpansionsviatranscorrelation. The Journal of Chemical Physics, 161(8):084104, 08
- [21]
-
[22]
Liguo Kong, Florian A. Bischoff, and Edward F. Valeev. Explicitly correlated r12/f12 methods for electronic structure.Chemical Reviews, 112(1):75– 107, 2012. doi: 10.1021/cr200204r
-
[23]
Amir Karton and Jan M. L. Martin. Explicitly cor- related wntheory: W1-f12 and w2-f12.The Jour- nal of Chemical Physics, 136(12):124114, 2012. doi: 10.1063/1.3697678
-
[24]
Peterson, Amir Karton, and Jan M
Nitai Sylvetsky, Kirk A. Peterson, Amir Karton, and Jan M. L. Martin. Toward a w4-f12 approach: Can explicitly correlated and orbital-basedab ini- tioccsd(t) limits be reconciled?The Journal of Chemical Physics, 144(21):214101, 2016. doi: 10.1063/1.4952410
-
[25]
Robert A. Shaw and J. Grant Hill. Approach- ing the hartree–fock limit through the comple- mentary auxiliary basis set singles correction and auxiliary basis sets.Journal of Chemical The- ory and Computation, 13(4):1691–1698, 2017. doi: 10.1021/acs.jctc.7b00140
-
[26]
Adler, Gerald Knizia, and Hans- Joachim Werner
Thomas B. Adler, Gerald Knizia, and Hans- Joachim Werner. A simple and efficient ccsd(t)-f12 approximation.The Journal of Chemical Physics, 127(22):221106, 2007. doi: 10.1063/1.2817618
-
[27]
Jozef Noga and Ján Šimunek. On the one- particle basis set relaxation in r12 based theo- ries.Chemical Physics, 356(1–3):1–6, 2009. doi: 10.1016/j.chemphys.2008.10.012
-
[28]
Attila Tajti, Péter G. Szalay, Attila G. Császár, Mihály Kállay, Jürgen Gauss, Edward F. Valeev, Bradley A. Flowers, Juana Vázquez, and John F. Stanton. Heat: High accuracy extrapolatedab 7 initiothermochemistry.The Journal of Chem- ical Physics, 121(23):11599–11613, 2004. doi: 10.1063/1.1811608
-
[29]
N. D. Drummond, M. D. Towler, and R. J. Needs. Jastrowcorrelationfactorforatoms, molecules, and solids.Physical Review B, 70(23):235119, 2004. doi: 10.1103/PhysRevB.70.235119
-
[30]
Ke Liao, Thomas Schraivogel, Hongjun Luo, Daniel Kats, and Ali Alavi. Towards efficient and accurate ab initio solutions to periodic systems via transcor- relation and coupled cluster theory.Phys. Rev. Res., 3:033072, Jul 2021. doi: 10.1103/PhysRevRe- search.3.033072
-
[31]
Osamu Hino, Yoshitaka Tanimura, and Seiichiro Ten-no. Biorthogonal approach for explicitly corre- lated calculations using the transcorrelated hamil- tonian.The Journal of Chemical Physics, 115(17): 7865–7871, 2001. doi: 10.1063/1.1404575
-
[32]
Curtiss, Krishnan Raghavachari, Gary W
Larry A. Curtiss, Krishnan Raghavachari, Gary W. Trucks, and John A. Pople. Gaussian-2 theory for molecular energies of first- and second-row com- pounds.The Journal of Chemical Physics, 94(11): 7221–7230, 1991. doi: 10.1063/1.460205
-
[33]
Yuan Yao, Emmanuel Giner, Junhao Li, Julien Toulouse, and Cyrus J. Umrigar. Almost exact en- ergies for the gaussian-2 set with the semistochas- ticheat-bathconfigurationinteractionmethod.The Journal of Chemical Physics, 153(12):124117, 2020. doi: 10.1063/5.0018577
-
[34]
Jan Řezáč and Pavel Hobza. Describing noncova- lent interactions beyond the common approxima- tions: How accurate is the “gold standard,” ccsd(t) at the complete basis set limit?Journal of Chemi- cal Theory and Computation, 9(5):2151–2155, 2013. doi: 10.1021/ct400057w
-
[35]
Extensions and applications of the a24 data set of accurate interaction energies.Phys- ical Chemistry Chemical Physics, 17:19268–19277,
Jan Řezáč, Matúš Dubecký, Petr Jurečka, and Pavel Hobza. Extensions and applications of the a24 data set of accurate interaction energies.Phys- ical Chemistry Chemical Physics, 17:19268–19277,
-
[36]
doi: 10.1039/C5CP03151F. 8
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.