Explainable AI for Cancer Drug Response Prediction: Beyond Univariate Feature Attributions
Pith reviewed 2026-07-02 15:39 UTC · model grok-4.3
The pith
ILLUME+ is a post-hoc framework that generates multi-form explanations for AI models predicting cancer drug responses from gene profiles, going beyond single-gene scores.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
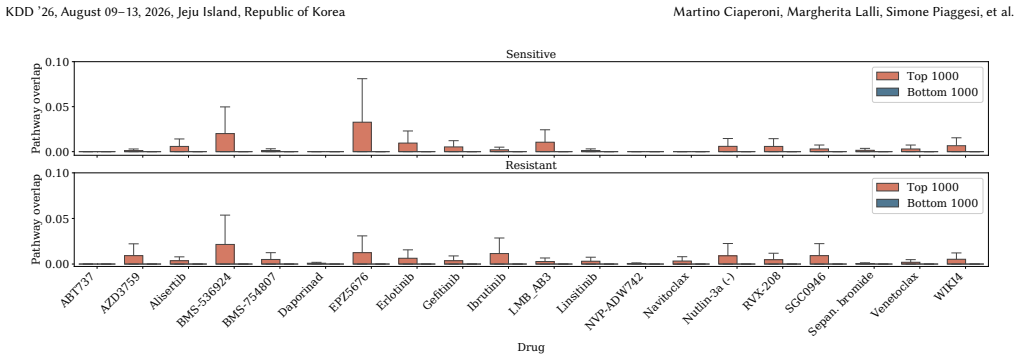
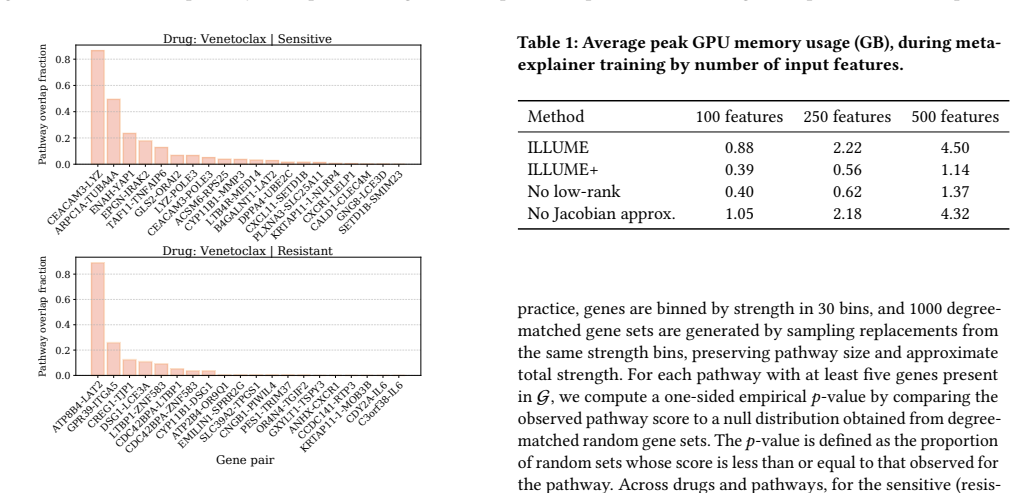
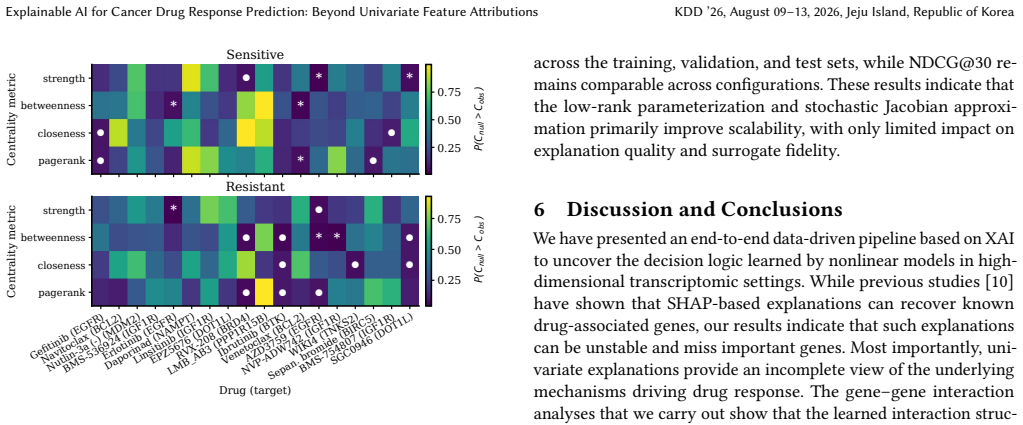
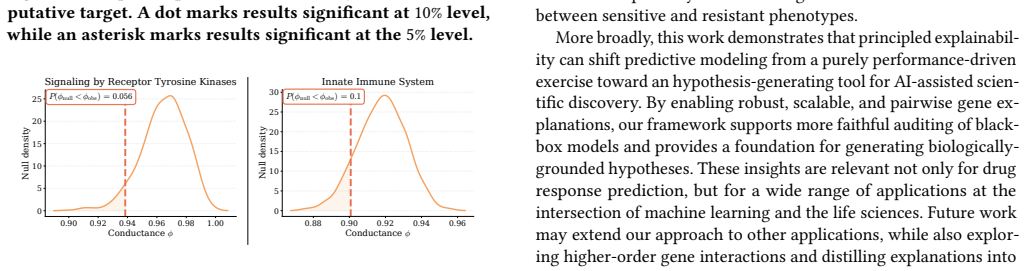
ILLUME+ is a scalable post-hoc explainability framework that moves beyond single-gene assessments to capture multiple, complementary forms of explanation. Integrated into an end-to-end pipeline for cancer drug response prediction from transcriptomic profiles, it produces more stable gene importance scores than existing baselines, recovers established drug-gene associations and mechanisms of action, and enables AI-assisted hypothesis generation to uncover novel interaction-driven molecular signals in cancer biology.
What carries the argument
ILLUME+, the post-hoc explainability framework that integrates multiple complementary explanation types to capture coordinated gene activity rather than univariate attributions.
If this is right
- Gene importance scores remain consistent when models are retrained or data splits change.
- Explanations recover known drug targets and mechanisms of action documented in cancer biology.
- AI outputs can be used to propose new hypotheses about gene interaction effects on drug sensitivity.
- The framework scales to large transcriptomic datasets without the computational limits of prior methods.
Where Pith is reading between the lines
- The same multi-explanation approach could transfer to other high-dimensional biological tasks such as predicting protein interactions or disease subtypes.
- If the novel signals hold up in follow-up experiments, the method could shorten the cycle from computational prediction to lab validation of drug mechanisms.
- Comparing ILLUME+ outputs against purely statistical gene correlation methods would clarify how much the AI model itself adds beyond data patterns.
Load-bearing premise
The post-hoc explanations generated by ILLUME+ accurately capture coordinated biological mechanisms rather than model-specific artifacts or data biases.
What would settle it
Re-running ILLUME+ on the same trained models and data yields gene importance scores that vary substantially across trials, or the recovered associations fail to match independently verified drug-gene relationships from the literature.
Figures


read the original abstract
Predicting cancer drug response from transcriptomic profiles is a cornerstone of precision oncology, yet the scientific value of machine learning models hinges not solely on predictive accuracy, but also on their capacity to generate reliable biological insights. Current explainability approaches in this setting are computationally costly, lack robustness, and reduce complex drug response to univariate gene importance scores, overlooking the coordinated gene activity that drives sensitivity and resistance. In this work, we present ILLUME+, a scalable post-hoc explainability framework that moves beyond single-gene assessments to capture multiple, complementary forms of explanation. Integrated into our end-to-end pipeline, ILLUME+ produces more stable gene importance scores than existing baselines, recovers established drug-gene associations and mechanisms of action, and enables AI-assisted hypothesis generation to uncover novel interaction-driven molecular signals in cancer biology.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper introduces ILLUME+, a scalable post-hoc explainability framework integrated into an end-to-end pipeline for predicting cancer drug response from transcriptomic profiles. It claims to move beyond univariate gene importance by capturing multiple complementary explanations, yielding more stable gene importance scores than baselines, recovering known drug-gene associations and mechanisms of action, and supporting AI-assisted hypothesis generation for novel interaction-driven molecular signals.
Significance. If the empirical claims are substantiated, the framework could meaningfully advance XAI applications in precision oncology by addressing limitations of current methods (computational cost, lack of robustness, and reduction to single-gene scores) and enabling more reliable extraction of coordinated biological insights from ML models.
major comments (2)
- [Abstract] Abstract: The abstract makes specific performance claims (more stable gene importance scores, recovery of established associations, enabling novel signal discovery) but provides no description of methods, datasets, baselines, stability metrics, validation procedures, or experimental design. Without these details the central claims cannot be evaluated for support.
- [Abstract] Abstract: No evidence or tests are supplied to address the risk that the generated explanations reflect model artifacts or data biases rather than coordinated biological mechanisms, leaving the claim of biological fidelity unsupported by the supplied text.
Simulated Author's Rebuttal
We thank the referee for their comments. We address the two major points on the abstract below, noting that the full manuscript provides the supporting details while agreeing that the abstract could be strengthened for standalone clarity.
read point-by-point responses
-
Referee: [Abstract] Abstract: The abstract makes specific performance claims (more stable gene importance scores, recovery of established associations, enabling novel signal discovery) but provides no description of methods, datasets, baselines, stability metrics, validation procedures, or experimental design. Without these details the central claims cannot be evaluated for support.
Authors: We agree the abstract is high-level and omits these specifics due to length constraints. The Methods section details the end-to-end pipeline, datasets (GDSC, CCLE, TCGA), baselines (SHAP, LIME, Integrated Gradients), stability metrics (e.g., Jaccard consistency across bootstrap runs and perturbations), and experimental design (cross-validation, known association recovery). We will revise the abstract to briefly reference the key datasets, baselines, and stability evaluation approach. revision: yes
-
Referee: [Abstract] Abstract: No evidence or tests are supplied to address the risk that the generated explanations reflect model artifacts or data biases rather than coordinated biological mechanisms, leaving the claim of biological fidelity unsupported by the supplied text.
Authors: The abstract summarizes the claims, but the full manuscript supports biological fidelity through recovery of established drug-gene associations and mechanisms of action (detailed in Results), plus stability comparisons. We acknowledge that explicit controls for artifacts (e.g., permutation baselines or bias audits) are not highlighted in the abstract. We will add a short discussion of these controls in the revised manuscript's Discussion section. revision: partial
Circularity Check
No derivation chain or equations present; no circularity identified
full rationale
The manuscript abstract and context describe an empirical XAI framework (ILLUME+) for post-hoc explanations in cancer drug response models. No equations, derivations, fitted parameters presented as predictions, or self-citation load-bearing uniqueness theorems appear in the supplied text. Claims rest on stability metrics, recovery of known associations, and hypothesis generation, which are externally falsifiable via experiments rather than reducing to self-definition or input renaming. The derivation chain is therefore self-contained with no detectable circular steps.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
2015.Foundations of linear and generalized linear models
Alan Agresti. 2015.Foundations of linear and generalized linear models. John Wiley & Sons
2015
-
[2]
Takuya Akiba et al. 2019. Optuna: A next-generation hyperparameter optimiza- tion framework. InACM SIGKDD international conference on knowledge discovery & data mining. 2623–2631
2019
-
[3]
Aoula Al-Zebeeby et al. 2018. Targeting intermediary metabolism enhances the efficacy of BH3 mimetic therapy in hematologic malignancies.Haematologica 104, 5 (2018), 1016
2018
-
[4]
Jordi Barretina et al. 2012. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity.Nature483, 7391 (2012), 603–607
2012
-
[5]
H Can Barutcu et al. 2025. Explainable artificial intelligence-based approaches for climate change: a review.International Journal of Global Warming35, 2-4 (2025), 244–260
2025
-
[6]
Iwona Bednarz-Misa et al. 2020. Interleukins 4 and 13 and their receptors are differently expressed in gastrointestinal tract cancers, depending on the anatomi- cal site and disease advancement, and improve colon cancer cell viability and motility.Cancers12, 6 (2020), 1463
2020
-
[7]
Elisabetta Botti et al. 2011. Developmental factor IRF6 exhibits tumor suppressor activity in squamous cell carcinomas.Proceedings of the National Academy of Sciences108, 33 (2011), 13710–13715
2011
-
[8]
Leo Breiman. 2001. Random Forests.Mach. Learn.45, 1 (2001), 5–32
2001
-
[9]
Aishwarya Budhkar et al. 2025. Demystifying the black box: A survey on explain- able artificial intelligence (XAI) in bioinformatics.Computational and Structural Biotechnology Journal(2025)
2025
-
[10]
Francesco Carli et al. 2025. Learning and actioning general principles of cancer cell drug sensitivity. 16, 1 (2025), 1654
2025
-
[11]
Jinyu Chen and Louxin Zhang. 2021. A survey and systematic assessment of computational methods for drug response prediction.Briefings in bioinformatics 22, 1 (2021), 232–246
2021
-
[12]
Steven M Corsello et al . 2020. Discovering the anticancer potential of non- oncology drugs by systematic viability profiling.Nature cancer1, 2 (2020), 235–248
2020
-
[13]
Beatriz Costa and Petia Georgieva. 2023. Explainable Artificial Intelligence in Healthcare Applications: A Systematic Review. In2023 International Scientific Conference on Computer Science (COMSCI). 1–8
2023
-
[14]
Liyuan Cui et al. 2024. GIMAP7 inhibits epithelial-mesenchymal transition and glycolysis in lung adenocarcinoma cells via regulating the Smo/AMPK signaling pathway.Thoracic Cancer15, 4 (2024), 286–298
2024
-
[15]
Janez Demsar. 2006. Statistical Comparisons of Classifiers over Multiple Data Sets.J. Mach. Learn. Res.7 (2006), 1–30
2006
-
[16]
Sarah C DiDonna et al. 2023. P4HTM: a novel downstream target of GATA3 in breast cancer.Research Square(2023), rs–3
2023
-
[17]
Linton C Freeman. 1978. Centrality in social networks conceptual clarification. Social networks1, 3 (1978), 215–239
1978
-
[18]
Judyta Gorka et al . 2021. MCPIP1 inhibits Wnt/ 𝛽-catenin signaling pathway activity and modulates epithelial-mesenchymal transition during clear cell renal cell carcinoma progression by targeting miRNAs.Oncogene40, 50 (2021), 6720– 6735
2021
-
[19]
Riccardo Guidotti et al . 2018. A survey of methods for explaining black box models.ACM computing surveys51, 5 (2018), 1–42
2018
-
[20]
Riccardo Guidotti et al. 2024. Stable and actionable explanations of black-box models through factual and counterfactual rules.Data Min. Knowl. Discov.38, 5 (2024), 2825–2862
2024
-
[21]
David Ha et al. 2017. HyperNetworks. InICLR (Poster). OpenReview.net
2017
- [22]
-
[23]
Noah Hollmann, Samuel Müller, Lennart Purucker, Arjun Krishnakumar, Max Körfer, Shi Bin Hoo, Robin Tibor Schirrmeister, and Frank Hutter. 2025. Accurate predictions on small data with a tabular foundation model.Nat.637, 8044 (2025), 319–326
2025
-
[24]
Wen Hwang et al. 2017. Expression of neuroendocrine factor VGF in lung cancer cells confers resistance to EGFR kinase inhibitors and triggers epithelial-to- mesenchymal transition.Cancer research77, 11 (2017), 3013–3026
2017
-
[25]
Francesco Iorio et al. 2016. A landscape of pharmacogenomic interactions in cancer.Cell166, 3 (2016), 740–754
2016
-
[26]
Janizek et al
Joseph D. Janizek et al. 2023. Uncovering expression signatures of synergistic drug responses via ensembles of explainable machine-learning models.Nature Biomedical Engineering7, 6 (2023), 811–829
2023
-
[27]
Kalervo Järvelin and Jaana Kekäläinen. 2002. Cumulated gain-based evaluation of IR techniques.ACM Transactions on Information Systems20, 4 (2002), 422–446
2002
-
[28]
Tasnuva D Kabir et al. 2025. Inhibition of the Caveolin-1 pathway promotes apop- tosis and overcomes pan-tyrosine kinase inhibitor resistance in hepatocellular carcinoma.Cell Death & Disease16, 1 (2025), 561
2025
-
[29]
Guolin Ke et al. 2017. LightGBM: A Highly Efficient Gradient Boosting Decision Tree. InNIPS. 3146–3154
2017
-
[30]
Philip Keyl et al. 2025. Neural interaction explainable AI predicts drug response across cancers.NAR cancer7, 3 (2025), zcaf029
2025
-
[31]
Simon Kind et al. 2024. KLK7 expression in human tumors: a tissue microarray study on 13,447 tumors.BMC cancer24, 1 (2024), 794
2024
-
[32]
Brent M Kuenzi et al. 2020. Predicting drug response and synergy using a deep learning model of human cancer cells.Cancer cell38, 5 (2020), 672–684
2020
-
[33]
Ritika Kundra et al. 2021. OncoTree: a cancer classification system for precision oncology.JCO clinical cancer informatics5 (2021), 221–230
2021
-
[34]
Miron B Kursa and Witold R Rudnicki. 2010. Feature selection with the Boruta package.Journal of statistical software36 (2010), 1–13
2010
-
[35]
Jingjing Li et al. 2023. Prognostic role of E2F1 gene expression in human cancer: a meta-analysis.BMC cancer23, 1 (2023), 509
2023
-
[36]
Weiquan Li et al. 2022. M2-polarization-related CNTNAP1 gene might be a novel immunotherapeutic target and biomarker for clear cell renal cell carcinoma. IUBMB life74, 5 (2022), 391–407
2022
-
[37]
Yang Lu et al. 2007. Epidermal growth factor receptor (EGFR) ubiquitination as a mechanism of acquired resistance escaping treatment by the anti-EGFR monoclonal antibody cetuximab.Cancer research67, 17 (2007), 8240–8247
2007
-
[38]
Scott M Lundberg et al. 2020. From local explanations to global understanding with explainable AI for trees.Nature machine intelligence2, 1 (2020), 56–67
2020
-
[39]
Lundberg and Su-In Lee
Scott M. Lundberg and Su-In Lee. 2017. A Unified Approach to Interpreting Model Predictions. InNIPS. 4765–4774
2017
-
[40]
Selma El Messaoudi-Aubert et al. 2010. Role for the MOV10 RNA helicase in polycomb-mediated repression of the INK4a tumor suppressor.Nature structural & molecular biology17, 7 (2010), 862–868
2010
-
[41]
Marija Milacic et al. 2024. The reactome pathway knowledgebase 2024.Nucleic acids research52, D1 (2024), D672–D678
2024
-
[42]
Patrick Mucka et al. 2023. CLK2 and CLK4 are regulators of DNA damage-induced NF-kB targeted by novel small molecule inhibitors.Cell Chemical Biology30, 10 (2023)
2023
-
[43]
Emilie Bousquet Mur et al . 2020. Notch inhibition overcomes resistance to tyrosine kinase inhibitors in EGFR-driven lung adenocarcinoma.The Journal of Clinical Investigation130, 2 (2020), 612–624
2020
-
[44]
Harshini Muralidharan et al. 2024. Breast Cancer stem cells upregulate IRF6 in stromal fibroblasts to induce stromagenesis.Cells13, 17 (2024), 1466
2024
-
[45]
Marco Padilla-Rodriguez et al. 2018. The actin cytoskeletal architecture of estro- gen receptor positive breast cancer cells suppresses invasion.Nature communi- cations9, 1 (2018), 2980
2018
-
[46]
Simone Piaggesi et al. 2025. Explanations Go Linear: Post-Hoc Explainability for Tabular Data with Interpretable Meta-Encoding. InICDM. IEEE, 663–672
2025
-
[47]
Joseph A Pinto et al. 2017. In silico evaluation of DNA Damage Inducible Tran- script 4 gene (DDIT4) as prognostic biomarker in several malignancies.Scientific reports7, 1 (2017), 1526
2017
-
[48]
Yan Qin et al. 2022. GIMAP7 as a potential predictive marker for pan-cancer prognosis and immunotherapy efficacy.Journal of inflammation research(2022), 1047–1061
2022
-
[49]
Matthew G Rees et al . 2016. Correlating chemical sensitivity and basal gene expression reveals mechanism of action.Nature chemical biology12, 2 (2016), 109–116
2016
-
[50]
Why should i trust you?
Marco Tulio Ribeiro et al. 2016. " Why should i trust you?" Explaining the pre- dictions of any classifier. InACM SIGKDD international conference on knowledge discovery and data mining. 1135–1144
2016
-
[51]
Rajat Rohatgi et al . 1999. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly.Cell97, 2 (1999), 221–231
1999
-
[52]
Alejandro Roisman et al. 2023. B4galt1 Regulates the WNT-𝛽-Catenin Axis to Control Hematopoietic Stem and Progenitor Cells (HSPCs) Fitness.Blood142 (2023), 398
2023
-
[53]
Tetsuroh Saitoh et al. 2001. Molecular cloning and characterization of FRAT2, encoding a positive regulator of the WNT signaling pathway.Biochemical and Biophysical Research Communications281, 3 (2001), 815–820
2001
-
[54]
Bikash Ranjan Samal et al. 2022. Opportunities and challenges in interpretable deep learning for drug sensitivity prediction of cancer cells.Frontiers in Bioinfor- matics2 (2022), 1036963
2022
-
[55]
Haoyuan Shi et al . 2025. DRExplainer: Quantifiable interpretability in drug response prediction with directed graph convolutional network.Artificial Intelli- gence in Medicine163 (2025), 103101. Explainable AI for Cancer Drug Response Prediction: Beyond Univariate Feature Attributions KDD ’26, August 09–13, 2026, Jeju Island, Republic of Korea
2025
-
[56]
Jingwei Shi et al. 2022. Loss of interleukin-13-receptor-alpha-1 induces apoptosis and promotes EMT in pancreatic cancer.International Journal of Molecular Sciences23, 7 (2022), 3659
2022
-
[57]
Sabbir Ahmed Sibli et al . 2025. Enhancing protein structure predictions: DeepSHAP as a tool for understanding AlphaFold2.Expert Systems with Applica- tions(2025), 127853
2025
-
[58]
Hongbin Su et al. 2021. Ubiquitin-like protein UBD promotes cell proliferation in colorectal cancer by facilitating p53 degradation.Frontiers in oncology11 (2021), 691347
2021
-
[59]
Aravind Subramanian et al. 2005. Gene set enrichment analysis: a knowledge- based approach for interpreting genome-wide expression profiles.Proceedings of the National Academy of Sciences102, 43 (2005), 15545–15550
2005
-
[60]
Hyuna Sung et al. 2021. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries.CA: a cancer journal for clinicians71, 3 (2021), 209–249
2021
-
[61]
Damian Szklarczyk et al. 2023. The STRING database in 2023: protein–protein association networks and functional enrichment analyses for any sequenced genome of interest.Nucleic acids research51, D1 (2023), D638–D646
2023
-
[62]
Yi-Ching Tang and Assaf Gottlieb. 2021. Explainable drug sensitivity prediction through cancer pathway enrichment.Scientific reports11, 1 (2021), 3128
2021
-
[63]
Christin Tse et al. 2008. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor.Cancer research68, 9 (2008), 3421–3428
2008
-
[64]
2011.Data mining and statistics for decision making
Stéphane Tufféry. 2011.Data mining and statistics for decision making. John Wiley & Sons
2011
-
[65]
Mathias Uhlén et al. 2015. Tissue-based map of the human proteome.Science 347, 6220 (2015), 1260419
2015
-
[66]
Mian Xie et al. 2013. Notch-1 contributes to epidermal growth factor receptor tyrosine kinase inhibitor acquired resistance in non-small cell lung cancer in vitro and in vivo.European journal of cancer49, 16 (2013), 3559–3572
2013
-
[67]
Li-Hao Yang et al. 2022. Neuronal survival factor VGF promotes chemoresis- tance and predicts poor prognosis in lung cancers with neuroendocrine feature. International Journal of Cancer151, 9 (2022), 1611–1625
2022
-
[68]
Jun Yin et al. 2019. let-7 and miR-17 promote self-renewal and drive gefitinib resistance in non-small cell lung cancer.Oncology Reports42, 2 (2019), 495–508
2019
-
[69]
Ming Yu et al. 2023. Elevated EVL methylation level in the normal colon mu- cosa is a potential risk biomarker for developing recurrent adenomas.Cancer Epidemiology, Biomarkers & Prevention32, 9 (2023), 1146–1152
2023
-
[70]
Qingbei Zeng et al. 2015. Discovery and evaluation of clinical candidate AZD3759, a potent, oral active, central nervous system-penetrant, epidermal growth factor receptor tyrosine kinase inhibitor.Journal of medicinal chemistry58, 20 (2015), 8200–8215
2015
-
[71]
Xiaoting Zhong et al. 2022. Explainable machine learning in materials science. npj computational materials8, 1 (2022), 204
2022
-
[72]
Jinfeng Zhu et al. 2022. FAT10 promotes chemotherapeutic resistance in pancre- atic cancer by inducing epithelial-mesenchymal transition via stabilization of FOXM1 expression.Cell death & disease13, 5 (2022), 497. APPENDIX A Data sources Transcriptomic data are derived from theCancer Cell Line Ency- clopedia(CCLE) [ 4], a collection of bulk RNA-seq data f...
2022
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.