An Additive MLP-GNN Framework for Characterizing Chemical and Structural Contributions to Aqueous Solubility
Pith reviewed 2026-07-03 05:43 UTC · model grok-4.3
The pith
An additive MLP-GNN model keeps chemical descriptors and molecular graph topology separate so their contributions to solubility can be inspected independently after training.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
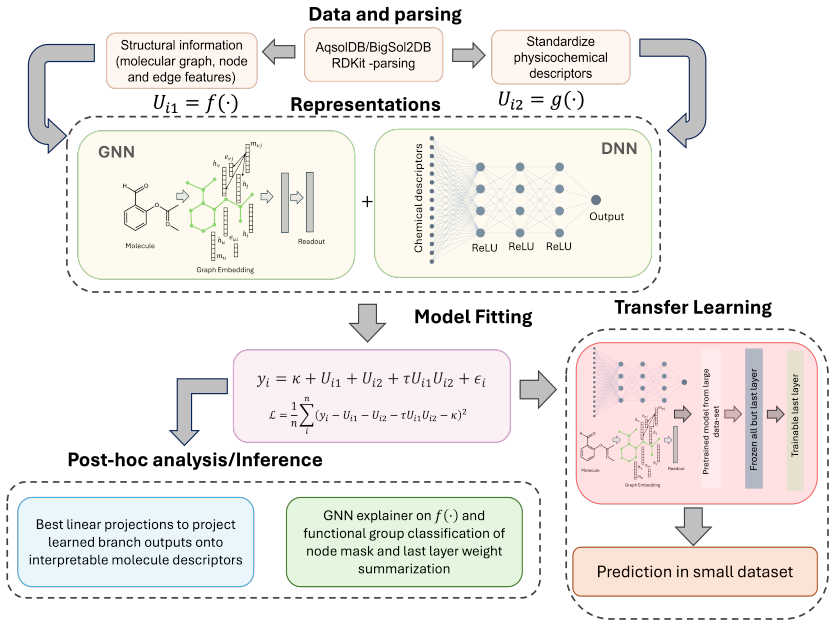
The central claim is that an additive combination of an MLP branch on chemical descriptors and a GNN branch on graph topology, with an optional multiplicative interaction, yields competitive solubility predictions while permitting direct post-training inspection of the separate chemical and structural contributions, with the separation preserved throughout training and further strengthened by pretraining on AqSolDB then fine-tuning on BigSolDB2.
What carries the argument
The additive model (with optional multiplicative interaction) that combines the MLP chemical-branch output and the GNN structural-branch output only at the prediction stage.
If this is right
- Chemical and structural components of each prediction can be examined separately after training.
- Pretraining on the larger AqSolDB dataset followed by fine-tuning on BigSolDB2 improves accuracy and reduces run-to-run variation.
- Best-linear-projection and embedding analyses show the chemical branch aligns with familiar physicochemical descriptors.
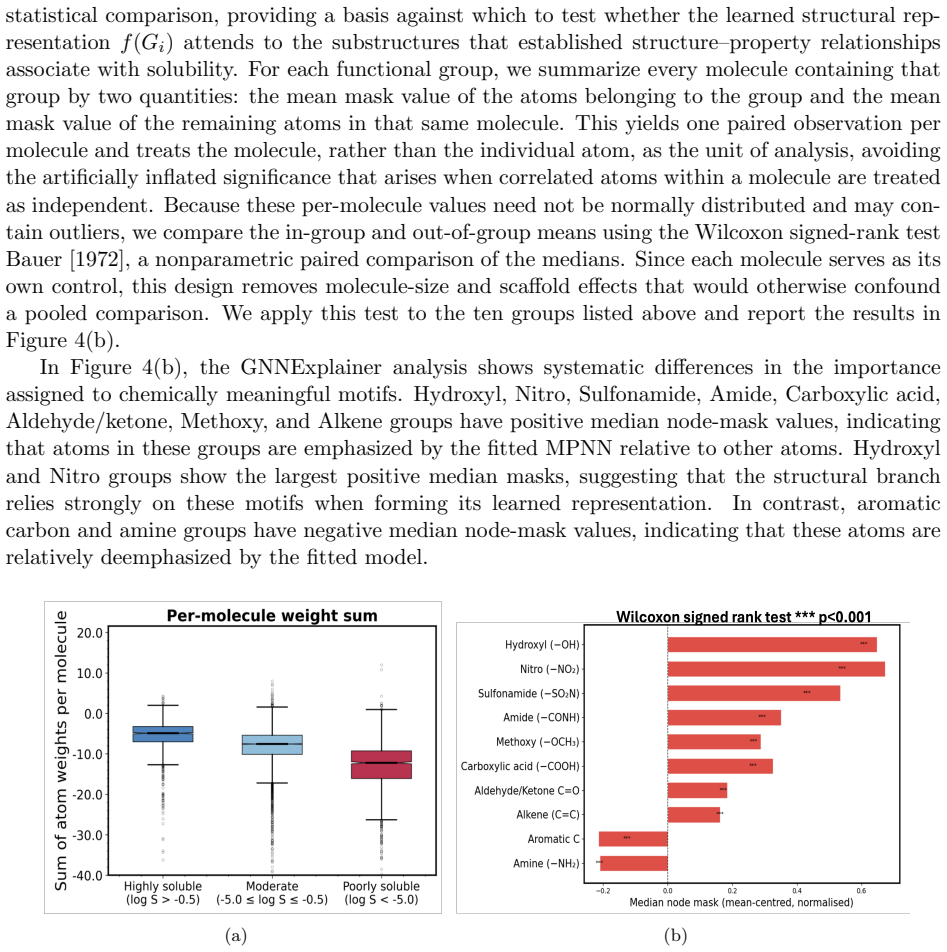
- GNNExplainer masks aggregated over functional groups show the structural branch captures graph-topological and functional-group patterns associated with solubility.
- The framework attains competitive predictive performance on both datasets while keeping the roles of the two information sources transparent.
Where Pith is reading between the lines
- If the additive decomposition remains stable, the same branch structure could be applied to other molecular properties where global chemistry and topology need to be distinguished.
- Drug-design workflows could query the separate branches to decide whether to modify molecular features or connectivity when attempting to adjust solubility.
- Repeating the pretrain-then-fine-tune protocol on additional solubility or related property datasets would test whether the learned features generalize beyond the two collections used here.
Load-bearing premise
That an additive combination of the two branch outputs is sufficient to capture their joint effect on solubility without requiring fusion steps that would destroy separability.
What would settle it
A head-to-head test in which a non-additive fused model achieves markedly higher accuracy on held-out solubility data while the additive version fails to produce clean, independent branch contributions.
Figures


read the original abstract
Aqueous solubility is a key property in early-stage drug discovery, but most predictive models merge physicochemical descriptors and molecular graph information into a single representation, obscuring whether a prediction is driven by global chemistry, molecular structure, or both. We present an additive deep-learning framework that keeps these two sources of information separate throughout training: physicochemical descriptors are encoded by a multilayer perceptron (the chemical branch) and molecular graph topology by a graph neural network (the structural branch), with the two outputs combined only at the prediction stage through an additive model with an optional multiplicative interaction. This design provides a direct decomposition of chemical and structural components that can be examined separately after training. Furthermore, pretraining on the larger AqSolDB dataset and fine-tuning on the smaller BigSolDB2 dataset substantially improve accuracy and reduce run-to-run variations, indicating generalizability of the learned features from the data-rich settings. We further interpret the fitted model using best linear projections of the branch outputs, molecule-level embedding summaries across solubility classes, and atom-level GNNExplainer masks aggregated over functional groups. These analyses show that the chemical branch aligns with familiar physicochemical descriptors, while the structural branch captures graph-topological and functional-group patterns associated with solubility. Across both datasets, the framework attains competitive predictive performance while making the distinct roles of chemical and structural information more transparent.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript proposes an additive MLP-GNN framework for aqueous solubility prediction in which physicochemical descriptors are processed by a multilayer perceptron (chemical branch) and molecular graph topology by a graph neural network (structural branch). The branch outputs are combined only at the final prediction stage via an additive model that optionally includes a multiplicative interaction term. This architecture is claimed to enable direct post-training decomposition of chemical versus structural contributions, supported by interpretability analyses (best linear projections, class-wise embedding summaries, and aggregated GNNExplainer masks). Pretraining on the larger AqSolDB dataset followed by fine-tuning on BigSolDB2 is reported to improve accuracy and reduce variance, yielding competitive predictive performance on both datasets.
Significance. If the reported empirical results and ablation studies hold, the work supplies a practical, separable architecture for cheminformatics tasks that require both accuracy and post-hoc attribution of distinct information sources. The pretraining/fine-tuning protocol and the alignment of learned branches with established physicochemical and functional-group patterns constitute concrete, reproducible strengths that could be adopted in related property-prediction settings.
minor comments (3)
- [Abstract] Abstract: the claim of 'competitive predictive performance' and 'substantially improve accuracy' is stated without any numerical values, baseline comparisons, or error bars; moving at least the headline metrics (e.g., RMSE or R² on the test splits) into the abstract would make the central empirical claim immediately verifiable.
- [Methods] The precise algebraic form of the optional multiplicative interaction term is not shown in the provided abstract; a short equation or pseudocode in §3 would clarify whether the interaction preserves the claimed separability of the two branches.
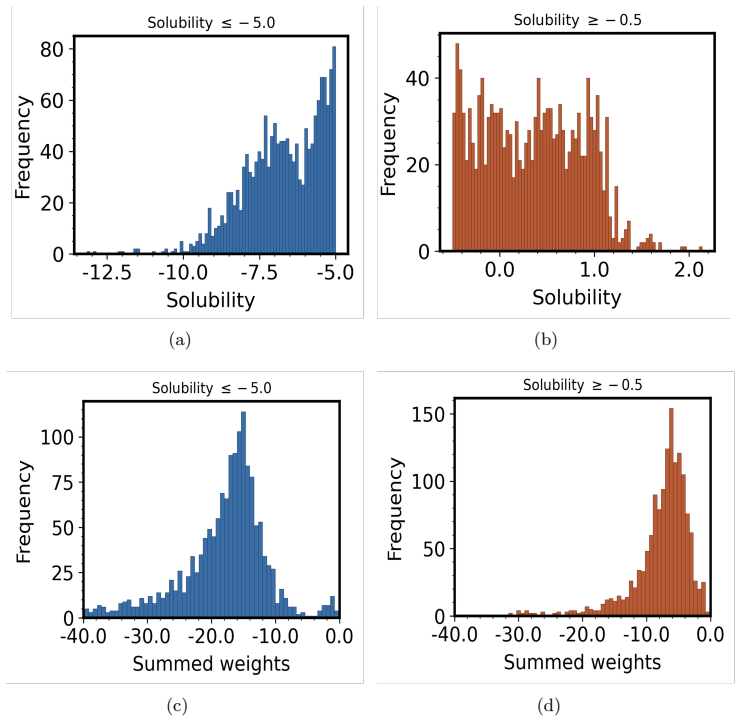
- [Results] Figure captions and axis labels for the embedding summaries and GNNExplainer masks should explicitly state the number of molecules or atoms aggregated and the statistical procedure used to obtain the reported patterns.
Simulated Author's Rebuttal
We thank the referee for the positive summary, significance assessment, and recommendation of minor revision. No major comments appear in the provided report, so we have no specific points requiring rebuttal or clarification at this stage.
Circularity Check
No significant circularity detected
full rationale
The paper presents an additive MLP-GNN architecture as an explicit modeling choice to enable separable decomposition of chemical (MLP) and structural (GNN) contributions, with outputs combined additively at the prediction stage. This separability is a direct consequence of the stated design rather than a derived result that reduces to fitted inputs or self-citations. No equations or claims in the abstract reduce predictions to parameters by construction, and the pretraining/fine-tuning protocol is a standard empirical transfer-learning step evaluated on accuracy and variance. Post-hoc interpretability methods (linear projections, embeddings, GNNExplainer) are enabled by the architecture but do not form a circular derivation chain. The framework's claims rest on independent design decisions and empirical results, making the derivation self-contained with no load-bearing self-referential steps.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Neural networks can be trained to produce branch outputs whose additive combination yields accurate predictions while preserving interpretability of each branch.
Reference graph
Works this paper leans on
-
[1]
AqSolDB, a curated reference set of aqueous solubility and 2D descriptors for a diverse set of compounds , author=. Scientific data , volume=. 2019 , publisher=
work page 2019
-
[2]
How Molecular Weight Shapes the Relative Importance of Chemical and Structural Descriptors for Aqueous Solubility , author=. 2025 , publisher=
work page 2025
-
[3]
Journal of the American Statistical Association , volume=
Constructing confidence sets using rank statistics , author=. Journal of the American Statistical Association , volume=. 1972 , publisher=
work page 1972
-
[4]
Mathematical Methods of Operations Research , volume=
An approximate subgradient algorithm for unconstrained nonsmooth, nonconvex optimization , author=. Mathematical Methods of Operations Research , volume=. 2008 , publisher=
work page 2008
-
[5]
Journal of Machine Learning Research , volume=
Benchmarking graph neural networks , author=. Journal of Machine Learning Research , volume=
-
[6]
The Econometrics Journal , volume=
Debiased machine learning of conditional average treatment effects and other causal functions , author=. The Econometrics Journal , volume=. 2021 , publisher=
work page 2021
-
[7]
Journal of the American Statistical Association , volume=
Distribution-free predictive inference for regression , author=. Journal of the American Statistical Association , volume=. 2018 , publisher=
work page 2018
-
[8]
SIAM Journal on Optimization , volume=
Convergence of the gradient sampling algorithm for nonsmooth nonconvex optimization , author=. SIAM Journal on Optimization , volume=. 2007 , publisher=
work page 2007
-
[9]
International conference on artificial intelligence and statistics , pages=
Linearly convergent Frank-Wolfe with backtracking line-search , author=. International conference on artificial intelligence and statistics , pages=. 2020 , organization=
work page 2020
-
[10]
Subgradient and sampling algorithms for l 1 regression , author=. Symposium on Discrete Algorithms: Proceedings of the sixteenth annual ACM-SIAM symposium on Discrete algorithms , volume=
-
[11]
lecture notes of EE392o, Stanford University, Autumn Quarter , volume=
Subgradient methods , author=. lecture notes of EE392o, Stanford University, Autumn Quarter , volume=
- [12]
-
[13]
AKCE International Journal of Graphs and Combinatorics , volume=
Global efficiency of graphs , author=. AKCE International Journal of Graphs and Combinatorics , volume=. 2015 , publisher=
work page 2015
-
[14]
Poor aqueous solubility—an industry wide problem in drug discovery , author=. Am Pharm Rev , volume=
-
[15]
Navigation Bar , author=. Navigation , volume=. 2018 , publisher=
work page 2018
-
[16]
Journal of pharmacological and toxicological methods , volume=
Drug-like properties and the causes of poor solubility and poor permeability , author=. Journal of pharmacological and toxicological methods , volume=. 2000 , publisher=
work page 2000
-
[17]
Annual review of pharmacology and toxicology , volume=
High-throughput screening in drug metabolism and pharmacokinetic support of drug discovery , author=. Annual review of pharmacology and toxicology , volume=. 2000 , publisher=
work page 2000
-
[18]
Molecular informatics , volume=
Application of random forest and multiple linear regression techniques to QSPR prediction of an aqueous solubility for military compounds , author=. Molecular informatics , volume=. 2010 , publisher=
work page 2010
-
[19]
Advanced drug delivery reviews , volume=
Prediction of drug solubility from structure , author=. Advanced drug delivery reviews , volume=. 2002 , publisher=
work page 2002
-
[20]
Molecular generative graph neural networks for drug discovery , author=. Neurocomputing , volume=. 2021 , publisher=
work page 2021
-
[21]
Drug Discovery Today , volume=
Graph neural networks for automated de novo drug design , author=. Drug Discovery Today , volume=. 2021 , publisher=
work page 2021
-
[22]
Global and local features of semantic networks: Evidence from the Hebrew mental lexicon , author=. PloS one , volume=. 2011 , publisher=
work page 2011
-
[23]
Journal of machine learning research , volume=
Sparse Bayesian learning and the relevance vector machine , author=. Journal of machine learning research , volume=
-
[24]
A new measure of centrality for brain networks , author=. PloS one , volume=. 2010 , publisher=
work page 2010
- [25]
-
[26]
Gábor Csárdi and Tamás Nepusz and Vincent Traag and Szabolcs Horvát and Fabio Zanini and Daniel Noom and Kirill Müller , year =
-
[27]
S-core network decomposition: A generalization of k-core analysis to weighted networks , author=. Physical Review E , volume=. 2013 , publisher=
work page 2013
-
[28]
Physica A: Statistical Mechanics and its Applications , volume=
The use of nodes attributes in social network analysis with an application to an international trade network , author=. Physica A: Statistical Mechanics and its Applications , volume=. 2018 , publisher=
work page 2018
-
[29]
Proceedings of the Twenty-Seventh Conference on Uncertainty in Artificial Intelligence , pages=
Modeling social networks with node attributes using the multiplicative attribute graph model , author=. Proceedings of the Twenty-Seventh Conference on Uncertainty in Artificial Intelligence , pages=
-
[30]
Journal of cheminformatics , volume=
Could graph neural networks learn better molecular representation for drug discovery? A comparison study of descriptor-based and graph-based models , author=. Journal of cheminformatics , volume=. 2021 , publisher=
work page 2021
-
[31]
PharmaNet: Pharmaceutical discovery with deep recurrent neural networks , author=. Plos one , volume=. 2021 , publisher=
work page 2021
-
[32]
Generating focused molecule libraries for drug discovery with recurrent neural networks , author=. ACS central science , volume=. 2018 , publisher=
work page 2018
-
[33]
Journal of chemical information and modeling , volume=
Virtual exploration of the chemical universe up to 11 atoms of C, N, O, F: assembly of 26.4 million structures (110.9 million stereoisomers) and analysis for new ring systems, stereochemistry, physicochemical properties, compound classes, and drug discovery , author=. Journal of chemical information and modeling , volume=. 2007 , publisher=
work page 2007
-
[34]
Journal of cheminformatics , volume=
A self-attention based message passing neural network for predicting molecular lipophilicity and aqueous solubility , author=. Journal of cheminformatics , volume=. 2020 , publisher=
work page 2020
-
[35]
Journal of the American Chemical Society , volume=
The first general index of molecular complexity , author=. Journal of the American Chemical Society , volume=. 1981 , publisher=
work page 1981
-
[36]
Journal of Molecular Graphics and Modelling , volume=
A widely applicable set of descriptors , author=. Journal of Molecular Graphics and Modelling , volume=. 2000 , publisher=
work page 2000
-
[37]
Journal of chemical information and computer sciences , volume=
ESOL: estimating aqueous solubility directly from molecular structure , author=. Journal of chemical information and computer sciences , volume=. 2004 , publisher=
work page 2004
-
[38]
Journal of Medicinal Chemistry , volume=
Hydrogen-bond donors in drug design , author=. Journal of Medicinal Chemistry , volume=. 2022 , publisher=
work page 2022
-
[39]
Bioorganic & medicinal chemistry , volume=
Predictive models of aqueous solubility of organic compounds built on A large dataset of high integrity , author=. Bioorganic & medicinal chemistry , volume=. 2019 , publisher=
work page 2019
-
[40]
Virtual ADMET assessment in target selection and maturation , volume=
Molecular fields to assess recognition forces and property spaces , author=. Virtual ADMET assessment in target selection and maturation , volume=. 2006 , publisher=
work page 2006
-
[41]
Computational Materials Science , volume=
Linking stability with molecular geometries of perovskites and lanthanide richness using machine learning methods , author=. Computational Materials Science , volume=. 2024 , publisher=
work page 2024
-
[42]
Nature Machine Intelligence , volume=
A geometric deep learning approach to predict binding conformations of bioactive molecules , author=. Nature Machine Intelligence , volume=. 2021 , publisher=
work page 2021
-
[43]
Journal of Cheminformatics , volume=
Prediction of organic compound aqueous solubility using machine learning: a comparison study of descriptor-based and fingerprints-based models , author=. Journal of Cheminformatics , volume=. 2023 , publisher=
work page 2023
-
[44]
Novel solubility prediction models: Molecular fingerprints and physicochemical features vs graph convolutional neural networks , author=. ACS omega , volume=. 2022 , publisher=
work page 2022
-
[45]
Journal of Chemical Information and Modeling , volume=
Blinded predictions and post hoc analysis of the second solubility challenge data: exploring training data and feature set selection for machine and deep learning models , author=. Journal of Chemical Information and Modeling , volume=. 2023 , publisher=
work page 2023
-
[46]
Journal of chemometrics , volume=
Machine learning in prediction of intrinsic aqueous solubility of drug-like compounds: Generalization, complexity, or predictive ability? , author=. Journal of chemometrics , volume=. 2021 , publisher=
work page 2021
-
[47]
Journal of chemical information and modeling , volume=
Deep architectures and deep learning in chemoinformatics: the prediction of aqueous solubility for drug-like molecules , author=. Journal of chemical information and modeling , volume=. 2013 , publisher=
work page 2013
-
[48]
and Katzberger, Paul and Maeder, Niels and Landrum, Gregory A
Lehner, Marc T. and Katzberger, Paul and Maeder, Niels and Landrum, Gregory A. and Riniker, Sereina , title =. The Journal of Chemical Physics , volume =
-
[49]
Journal of cheminformatics , volume=
One molecular fingerprint to rule them all: drugs, biomolecules, and the metabolome , author=. Journal of cheminformatics , volume=. 2020 , publisher=
work page 2020
-
[50]
Distance connectivity index , author=. Chem. Phys. Lett , volume=
-
[51]
Journal of the American chemical society , volume=
Structural determination of paraffin boiling points , author=. Journal of the American chemical society , volume=. 1947 , publisher=
work page 1947
-
[52]
Topological index. A newly proposed quantity characterizing the topological nature of structural isomers of saturated hydrocarbons , author=. Bulletin of the Chemical Society of Japan , volume=. 1971 , publisher=
work page 1971
-
[53]
Journal of the American Chemical Society , volume=
Characterization of molecular branching , author=. Journal of the American Chemical Society , volume=. 1975 , publisher=
work page 1975
-
[54]
Statistical modelling of molecular descriptors in QSAR/QSPR , author=. 2012 , publisher=
work page 2012
-
[55]
Drug discovery today , volume=
Machine learning in chemoinformatics and drug discovery , author=. Drug discovery today , volume=. 2018 , publisher=
work page 2018
-
[56]
International Journal of Molecular Sciences , volume=
Recent advances in machine-learning-based chemoinformatics: a comprehensive review , author=. International Journal of Molecular Sciences , volume=. 2023 , publisher=
work page 2023
- [57]
-
[58]
Nature Machine Intelligence , volume=
Molecular contrastive learning of representations via graph neural networks , author=. Nature Machine Intelligence , volume=. 2022 , publisher=
work page 2022
-
[59]
Journal of Cheminformatics , volume=
MolPROP: Molecular Property prediction with multimodal language and graph fusion , author=. Journal of Cheminformatics , volume=. 2024 , publisher=
work page 2024
-
[60]
Machine learning toxicity prediction: latest advances by toxicity end point , author=. ACS omega , volume=. 2022 , publisher=
work page 2022
-
[61]
Journal of the Royal Statistical Society Series C: Applied Statistics , volume=
Nonparametric group variable selection with multivariate response for connectome-based modelling of cognitive scores , author=. Journal of the Royal Statistical Society Series C: Applied Statistics , volume=. 2023 , publisher=
work page 2023
-
[62]
Frontiers in aging neuroscience , volume=
Feature selection and combination of information in the functional brain connectome for discrimination of mild cognitive impairment and analyses of altered brain patterns , author=. Frontiers in aging neuroscience , volume=. 2020 , publisher=
work page 2020
-
[63]
Can small drugs predict the intrinsic aqueous solubility of ‘beyond Rule of 5’big drugs? , author=. ADMET and DMPK , volume=. 2020 , publisher=
work page 2020
-
[64]
“Flexible-acceptor” general solubility equation for beyond rule of 5 drugs , author=. Molecular Pharmaceutics , volume=. 2020 , publisher=
work page 2020
-
[65]
Journal of medicinal chemistry , volume=
Practical high-quality electrostatic potential surfaces for drug discovery using a graph-convolutional deep neural network , author=. Journal of medicinal chemistry , volume=. 2019 , publisher=
work page 2019
-
[66]
Pedregosa, F. and Varoquaux, G. and Gramfort, A. and Michel, V. and Thirion, B. and Grisel, O. and Blondel, M. and Prettenhofer, P. and Weiss, R. and Dubourg, V. and Vanderplas, J. and Passos, A. and Cournapeau, D. and Brucher, M. and Perrot, M. and Duchesnay, E. , journal=. Scikit-learn: Machine Learning in
- [67]
-
[68]
Journal of Chemical Theory and Computation , volume=
Toward physics-based solubility computation for pharmaceuticals to rival informatics , author=. Journal of Chemical Theory and Computation , volume=. 2021 , publisher=
work page 2021
-
[69]
Biomolecular condensate microstructure is invariant to sequence-encoded molecular and macroscopic properties , author=. Soft Matter , year=
-
[70]
Nature Communications , volume=
Chemistry-intuitive explanation of graph neural networks for molecular property prediction with substructure masking , author=. Nature Communications , volume=. 2023 , publisher=
work page 2023
-
[71]
Deep Learning for the Life Sciences , author=
-
[72]
BigSolDB 2.0, dataset of solubility values for organic compounds in different solvents at various temperatures , author=. Scientific Data , volume=. 2025 , publisher=
work page 2025
-
[73]
Journal of chemical theory and computation , volume=
Graph neural networks for predicting solubility in diverse solvents using molmerger incorporating solute--solvent interactions , author=. Journal of chemical theory and computation , volume=. 2024 , publisher=
work page 2024
-
[74]
Journal of computer-aided molecular design , volume=
Molecular graph convolutions: moving beyond fingerprints , author=. Journal of computer-aided molecular design , volume=. 2016 , publisher=
work page 2016
-
[75]
International conference on machine learning , pages=
Neural message passing for quantum chemistry , author=. International conference on machine learning , pages=. 2017 , organization=
work page 2017
-
[76]
Advances in neural information processing systems , volume=
Gnnexplainer: Generating explanations for graph neural networks , author=. Advances in neural information processing systems , volume=
-
[77]
The properties of known drugs. 1. Molecular frameworks , author=. Journal of medicinal chemistry , volume=. 1996 , publisher=
work page 1996
- [78]
-
[79]
MoleculeNet: a benchmark for molecular machine learning , author=. Chemical science , volume=. 2018 , publisher=
work page 2018
-
[80]
Clinical Pharmacology & Therapeutics , volume =
Evaluating the role of solubility in oral absorption of poorly water-soluble drugs using physiologically-based pharmacokinetic modeling , author =. Clinical Pharmacology & Therapeutics , volume =. 2020 , doi =
work page 2020
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.