Doloris: Dual Conditional Diffusion Implicit Bridges with Sparsity Masking Strategy for Unpaired Single-Cell Perturbation Estimation
Pith reviewed 2026-05-19 08:00 UTC · model grok-4.3
The pith
Dual conditional diffusion models implicitly align unpaired control and perturbed single-cell distributions in a shared Gaussian latent space while using sparsity masking to preserve response diversity.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Doloris defines a new paradigm for modeling unpaired, high-dimensional, and sparse single-cell perturbation data. It leverages dual conditional diffusion models for separate learning of control and perturbed distributions, complemented by a sparsity masking strategy to enhance prediction of zero-valued genes.
What carries the argument
Dual conditional diffusion implicit bridges that map control and perturbed distributions into a shared Gaussian latent space without explicit cell pairing, together with a sparsity masking strategy that predicts zero-expressed genes so the diffusion models focus on meaningful non-zero patterns.
If this is right
- Perturbation effects can be predicted for individual cells without requiring paired pre- and post-perturbation measurements of the same cell.
- The sparsity mask allows the diffusion process to capture varied gene-expression patterns instead of defaulting to the dominant zero values in sparse data.
- State-of-the-art performance is achieved on public single-cell perturbation datasets by effectively modeling response diversity.
- Key genes can be identified and drug-screening efficiency improved through computational estimation rather than exhaustive wet-lab experiments.
Where Pith is reading between the lines
- The shared latent-space alignment may generalize to other unpaired biological modalities such as spatial transcriptomics where direct cell pairing is infeasible.
- Interpolating within the shared Gaussian space could enable prediction of cellular responses to novel or combined perturbations not seen in training.
- If the implicit bridge holds, similar dual-diffusion constructions might serve as distribution-matching tools for other unpaired high-dimensional settings outside single-cell biology.
Load-bearing premise
The control and perturbed distributions can be implicitly aligned through a shared Gaussian latent space without explicit cell pairing.
What would settle it
Running the model on a held-out single-cell dataset and finding that the generated perturbed profiles have significantly lower diversity or poorer match to the true perturbed distribution than competing methods, or that disabling the shared latent space alignment collapses performance to baseline levels.
Figures





read the original abstract
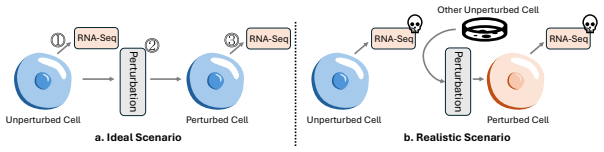
Estimating single-cell responses across various perturbations facilitates the identification of key genes and enhances drug screening, significantly boosting experimental efficiency. However, single-cell sequencing is a destructive process, making it impossible to capture the same cell's phenotype before and after perturbation. Consequently, data collected under perturbed and unperturbed conditions are inherently unpaired, creating a critical yet unresolved problem in single-cell perturbation modeling. Moreover, the high dimensionality and sparsity of single-cell expression make direct modeling prone to focusing on zeros and neglecting meaningful patterns. To address these problems, we propose a new paradigm for single-cell perturbation modeling. Specifically, we leverage dual diffusion models to learn the control and perturbed distributions separately, and implicitly align them through a shared Gaussian latent space, without requiring explicit cell pairing. Furthermore, we introduce a sparsity masking strategy in which the mask model learns to predict zero-expressed genes, allowing the diffusion model to focus on capturing meaningful patterns among expressed genes and thereby preserving diversity in high-dimensional sparse data. We introduce \textbf{Doloris}, a generative framework that defines a new paradigm for modeling unpaired, high-dimensional, and sparse single-cell perturbation data. It leverages dual conditional diffusion models for separate learning of control and perturbed distributions, complemented by a sparsity masking strategy to enhance prediction of zero-valued genes. The results on publicly available datasets show that our model effectively captures the diversity of single-cell perturbations and achieves state-of-the-art performance. To facilitate reproducibility, we include the code in the supplementary materials.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript introduces Doloris, a generative framework for unpaired single-cell perturbation estimation. It employs dual conditional diffusion models to separately learn the distributions of control and perturbed cells, implicitly aligning them through a shared Gaussian latent space without explicit cell pairing. A sparsity masking strategy is proposed where a mask model predicts zero-expressed genes to allow the diffusion model to focus on meaningful patterns. The authors report that the model captures the diversity of single-cell perturbations and achieves state-of-the-art performance on publicly available datasets, with code included for reproducibility.
Significance. This work has potential significance in advancing single-cell perturbation modeling by addressing the unpaired nature of data and the challenges of sparsity in scRNA-seq. If the implicit alignment successfully preserves biological perturbation effects, it could facilitate more efficient drug screening and key gene identification. The inclusion of reproducible code is a strength that supports the assessment.
major comments (2)
- [Method section on dual conditional diffusion implicit bridges] The central mechanism of implicit alignment through the shared Gaussian latent space (described in the dual conditional diffusion implicit bridges section) is not sufficiently validated for preserving cell-specific perturbation effects. The skeptic's concern lands: without explicit tests (e.g., on subsets with known pairings or using biological priors), it is possible that the generated perturbations match overall statistics but fail to reflect actual response diversity on the underlying cell population. This is load-bearing for the claim of effective modeling of unpaired data.
- [Results section] Table reporting main results: while SOTA performance is claimed, the manuscript should provide more detailed error analysis, variance across runs, and comparison to recent baselines in the field to substantiate the improvement, particularly regarding diversity metrics.
minor comments (2)
- [Abstract] The abstract could benefit from specifying the exact datasets and quantitative metrics used to claim SOTA performance.
- [Figure captions] Some figures illustrating the sparsity masking could have more detailed explanations of how it affects the diffusion process and diversity preservation.
Simulated Author's Rebuttal
We thank the referee for the constructive comments on our manuscript. We have addressed the concerns regarding validation of the implicit alignment mechanism and the presentation of experimental results by adding new analyses and details in the revised version.
read point-by-point responses
-
Referee: [Method section on dual conditional diffusion implicit bridges] The central mechanism of implicit alignment through the shared Gaussian latent space (described in the dual conditional diffusion implicit bridges section) is not sufficiently validated for preserving cell-specific perturbation effects. The skeptic's concern lands: without explicit tests (e.g., on subsets with known pairings or using biological priors), it is possible that the generated perturbations match overall statistics but fail to reflect actual response diversity on the underlying cell population. This is load-bearing for the claim of effective modeling of unpaired data.
Authors: We agree that explicit validation of cell-specific preservation is important for substantiating the implicit alignment. In the revised manuscript, we have added a new subsection under Methods describing experiments on a synthetic dataset constructed from known cell populations with simulated perturbations, allowing access to ground-truth pairings. We evaluate per-cell correlation between generated and true perturbed states, as well as preservation of known biological pathways as priors. These results, now included in the revised Results section with a supporting figure, show that the shared latent space captures individual cell responses beyond aggregate statistics. revision: yes
-
Referee: [Results section] Table reporting main results: while SOTA performance is claimed, the manuscript should provide more detailed error analysis, variance across runs, and comparison to recent baselines in the field to substantiate the improvement, particularly regarding diversity metrics.
Authors: We appreciate this suggestion to strengthen the empirical claims. The revised manuscript now includes an expanded main results table with mean performance and standard deviations computed over five independent runs. We have added a dedicated error analysis subsection reporting per-gene and per-cell error distributions. For diversity, we include additional metrics such as response entropy and unique gene count variance, along with comparisons to two recent baselines in the single-cell perturbation literature. These updates appear in the revised Table 1 and accompanying text. revision: yes
Circularity Check
Standard diffusion machinery with domain assumptions; no load-bearing circularity
full rationale
The paper proposes Doloris as a generative framework using dual conditional diffusion models to separately learn control and perturbed distributions, implicitly aligned via a shared Gaussian latent space, plus a sparsity masking strategy for high-dimensional sparse scRNA-seq data. This is framed as a new paradigm for unpaired perturbation estimation, with empirical SOTA claims on public datasets. No equations or steps in the provided text reduce predictions to fitted inputs by construction, nor do self-citations form a load-bearing chain for the core claims. The implicit alignment is presented as a modeling choice/assumption rather than a derived necessity that loops back to itself. This qualifies as a normal non-circular proposal resting on standard diffusion techniques and domain knowledge.
Axiom & Free-Parameter Ledger
axioms (2)
- domain assumption Single-cell sequencing is destructive, so perturbed and unperturbed measurements are inherently unpaired.
- domain assumption High-dimensional single-cell expression data is sparse with many zero entries that must be handled separately from expressed genes.
Lean theorems connected to this paper
-
IndisputableMonolith/Foundation/RealityFromDistinction.leanreality_from_one_distinction unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
leverage dual diffusion models to learn the control and perturbed distributions separately, and implicitly align them through a shared Gaussian latent space, without requiring explicit cell pairing
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
sparsity masking strategy in which the mask model learns to predict zero-expressed genes
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
Britt Adamson, Thomas M Norman, Marco Jost, Min Y Cho, James K Nuñez, Yuwen Chen, Jacqueline E Villalta, Luke A Gilbert, Max A Horlbeck, Marco Y Hein, et al. A multiplexed single-cell crispr screening platform enables systematic dissection of the unfolded protein response. Cell, 167(7):1867–1882, 2016
work page 2016
-
[2]
Applications of crispr technologies in research and beyond
Rodolphe Barrangou and Jennifer A Doudna. Applications of crispr technologies in research and beyond. Nature biotechnology, 34(9):933–941, 2016
work page 2016
-
[3]
Modelling cellular perturbations with the sparse additive mechanism shift variational autoencoder
Michael Bereket and Theofanis Karaletsos. Modelling cellular perturbations with the sparse additive mechanism shift variational autoencoder. Advances in Neural Information Processing Systems, 36, 2024
work page 2024
-
[4]
How Attentive are Graph Attention Networks?
Shaked Brody, Uri Alon, and Eran Yahav. How attentive are graph attention networks? arXiv preprint arXiv:2105.14491, 2021
work page internal anchor Pith review Pith/arXiv arXiv 2021
-
[5]
Learning single- cell perturbation responses using neural optimal transport
Charlotte Bunne, Stefan G Stark, Gabriele Gut, Jacobo Sarabia Del Castillo, Mitch Levesque, Kjong-Van Lehmann, Lucas Pelkmans, Andreas Krause, and Gunnar Rätsch. Learning single- cell perturbation responses using neural optimal transport. Nature methods, 20(11):1759–1768, 2023
work page 2023
-
[6]
Yichuan Cao, Xiamiao Zhao, Songming Tang, Qun Jiang, Sijie Li, Siyu Li, and Shengquan Chen. scbutterfly: a versatile single-cell cross-modality translation method via dual-aligned variational autoencoders. Nature Communications, 15(1):2973, 2024
work page 2024
-
[7]
Changxi Chi, Jun Xia, Jingbo Zhou, Jiabei Cheng, Chang Yu, and Stan Z Li. Grape: Hetero- geneous graph representation learning for genetic perturbation with coding and non-coding biotype. arXiv preprint arXiv:2505.03853, 2025
-
[8]
scgpt: toward building a foundation model for single-cell multi-omics using generative ai
Haotian Cui, Chloe Wang, Hassaan Maan, Kuan Pang, Fengning Luo, Nan Duan, and Bo Wang. scgpt: toward building a foundation model for single-cell multi-omics using generative ai. Nature Methods, 21(8):1470–1480, 2024
work page 2024
-
[9]
AnimateDiff: Animate Your Personalized Text-to-Image Diffusion Models without Specific Tuning
Yuwei Guo, Ceyuan Yang, Anyi Rao, Zhengyang Liang, Yaohui Wang, Yu Qiao, Maneesh Agrawala, Dahua Lin, and Bo Dai. Animatediff: Animate your personalized text-to-image diffusion models without specific tuning. arXiv preprint arXiv:2307.04725, 2023
work page internal anchor Pith review Pith/arXiv arXiv 2023
-
[10]
Large-scale foundation model on single-cell transcriptomics
Minsheng Hao, Jing Gong, Xin Zeng, Chiming Liu, Yucheng Guo, Xingyi Cheng, Taifeng Wang, Jianzhu Ma, Xuegong Zhang, and Le Song. Large-scale foundation model on single-cell transcriptomics. Nature methods, 21(8):1481–1491, 2024
work page 2024
-
[11]
Squidiff: Predicting cellular development and responses to perturbations using a diffusion model
Siyu He, Yuefei Zhu, Daniel Naveed Tavakol, Haotian Ye, Yeh-Hsing Lao, Zixian Zhu, Cong Xu, Sharadha Chauhan, Guy Garty, Raju Tomer, et al. Squidiff: Predicting cellular development and responses to perturbations using a diffusion model. bioRxiv, pages 2024–11, 2024
work page 2024
-
[13]
Predicting cellular responses to novel drug perturbations at a single-cell resolution
Leon Hetzel, Simon Boehm, Niki Kilbertus, Stephan Günnemann, Fabian Theis, et al. Predicting cellular responses to novel drug perturbations at a single-cell resolution. Advances in Neural Information Processing Systems, 35:26711–26722, 2022
work page 2022
-
[14]
Delivering crispr: a review of the challenges and approaches
Christopher A Lino, Jason C Harper, James P Carney, and Jerilyn A Timlin. Delivering crispr: a review of the challenges and approaches. Drug delivery, 25(1):1234–1257, 2018
work page 2018
-
[15]
Learning interpretable cellular responses to complex perturbations in high-throughput screens
M Lotfollahi, AK Susmelj, and C De Donno. Learning interpretable cellular responses to complex perturbations in high-throughput screens. biorxiv. 2021. 2021.04. 14.439903. 10
work page 2021
-
[16]
scgen predicts single-cell perturbation responses
Mohammad Lotfollahi, F Alexander Wolf, and Fabian J Theis. scgen predicts single-cell perturbation responses. Nature methods, 16(8):715–721, 2019
work page 2019
-
[17]
Understanding diffusion models: A unified perspective,
Calvin Luo. Understanding diffusion models: A unified perspective. arXiv preprint arXiv:2208.11970, 2022
-
[18]
Mapping and quantifying mammalian transcriptomes by rna-seq
Ali Mortazavi, Brian A Williams, Kenneth McCue, Lorian Schaeffer, and Barbara Wold. Mapping and quantifying mammalian transcriptomes by rna-seq. Nature methods, 5(7):621– 628, 2008
work page 2008
-
[19]
Exploring genetic interaction manifolds constructed from rich single-cell phenotypes
Thomas M Norman, Max A Horlbeck, Joseph M Replogle, Alex Y Ge, Albert Xu, Marco Jost, Luke A Gilbert, and Jonathan S Weissman. Exploring genetic interaction manifolds constructed from rich single-cell phenotypes. Science, 365(6455):786–793, 2019
work page 2019
-
[20]
scperturb: harmonized single-cell perturbation data
Stefan Peidli, Tessa D Green, Ciyue Shen, Torsten Gross, Joseph Min, Samuele Garda, Bo Yuan, Linus J Schumacher, Jake P Taylor-King, Debora S Marks, et al. scperturb: harmonized single-cell perturbation data. Nature Methods, 21(3):531–540, 2024
work page 2024
-
[21]
Gears: Predicting transcriptional outcomes of novel multi-gene perturbations
Yusuf Roohani, Kexin Huang, and Jure Leskovec. Gears: Predicting transcriptional outcomes of novel multi-gene perturbations. BioRxiv, pages 2022–07, 2022
work page 2022
-
[22]
Denoising Diffusion Implicit Models
Jiaming Song, Chenlin Meng, and Stefano Ermon. Denoising diffusion implicit models. arXiv preprint arXiv:2010.02502, 2020
work page internal anchor Pith review Pith/arXiv arXiv 2010
-
[24]
Massively multiplex chemical transcriptomics at single-cell resolution
Sanjay R Srivatsan, José L McFaline-Figueroa, Vijay Ramani, Lauren Saunders, Junyue Cao, Jonathan Packer, Hannah A Pliner, Dana L Jackson, Riza M Daza, Lena Christiansen, et al. Massively multiplex chemical transcriptomics at single-cell resolution. Science, 367(6473): 45–51, 2020
work page 2020
-
[25]
Dual diffusion implicit bridges for image-to-image translation
Xuan Su, Jiaming Song, Chenlin Meng, and Stefano Ermon. Dual diffusion implicit bridges for image-to-image translation. arXiv preprint arXiv:2203.08382, 2022
-
[26]
Petar Veliˇckovi´c, Guillem Cucurull, Arantxa Casanova, Adriana Romero, Pietro Lio, and Yoshua Bengio. Graph attention networks. arXiv preprint arXiv:1710.10903, 2017
work page internal anchor Pith review Pith/arXiv arXiv 2017
-
[27]
Scanpy: large-scale single-cell gene expression data analysis
F Alexander Wolf, Philipp Angerer, and Fabian J Theis. Scanpy: large-scale single-cell gene expression data analysis. Genome biology, 19:1–5, 2018
work page 2018
-
[28]
Predicting cellular responses with variational causal inference and refined relational information
Yulun Wu, Robert A Barton, Zichen Wang, Vassilis N Ioannidis, Carlo De Donno, Layne C Price, Luis F V oloch, and George Karypis. Predicting cellular responses with variational causal inference and refined relational information. arXiv preprint arXiv:2210.00116, 2022
-
[29]
Mole-bert: Rethinking pre-training graph neural networks for molecules
Jun Xia, Chengshuai Zhao, Bozhen Hu, Zhangyang Gao, Cheng Tan, Yue Liu, Siyuan Li, and Stan Z Li. Mole-bert: Rethinking pre-training graph neural networks for molecules. 2023
work page 2023
-
[30]
Xiaodong Yang, Guole Liu, Guihai Feng, Dechao Bu, Pengfei Wang, Jie Jiang, Shubai Chen, Qinmeng Yang, Hefan Miao, Yiyang Zhang, et al. Genecompass: deciphering universal gene regulatory mechanisms with a knowledge-informed cross-species foundation model. Cell Research, 34(12):830–845, 2024
work page 2024
-
[31]
Uni-mol: A universal 3d molecular representation learning framework
Gengmo Zhou, Zhifeng Gao, Qiankun Ding, Hang Zheng, Hongteng Xu, Zhewei Wei, Linfeng Zhang, and Guolin Ke. Uni-mol: A universal 3d molecular representation learning framework. 2023. 11 A Mask Model In this section, we present the design rationale and architecture of the Mask Model. Given the high-dimensional and sparse nature of gene expression data, dire...
work page 2023
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.