Quantum-centric simulation of hydrogen abstraction by sample-based quantum diagonalization and entanglement forging
Pith reviewed 2026-05-18 23:29 UTC · model grok-4.3
The pith
Entanglement forging combined with sample-based quantum diagonalization computes activation and reaction energies for hydrogen abstraction on a superconducting processor.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
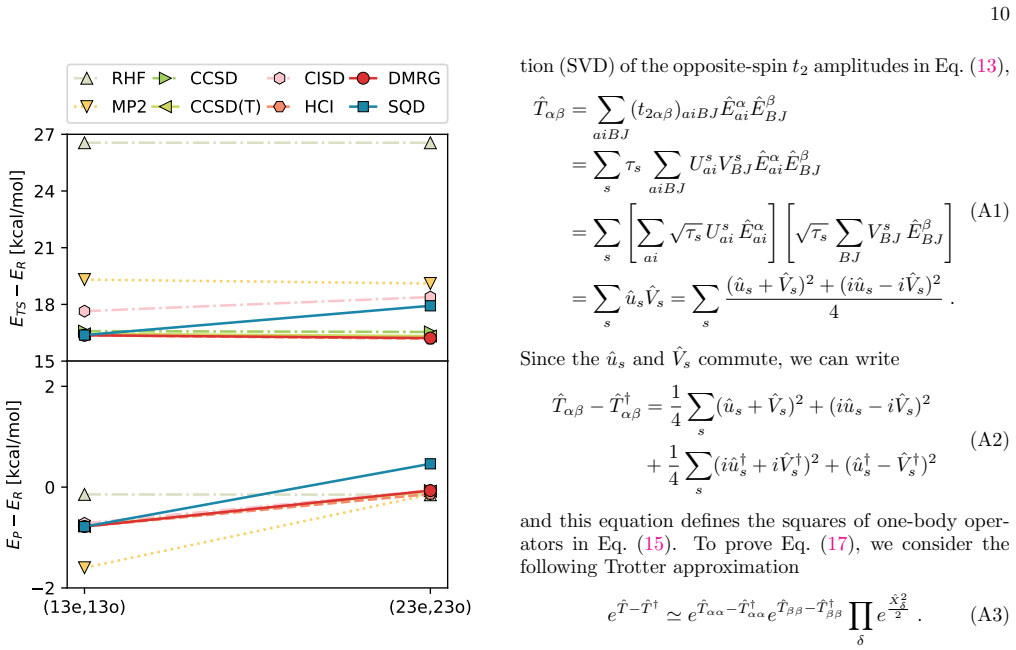
The authors establish that the combined EF-SQD approach, executed on a superconducting quantum processor, produces activation energies and reaction energies for hydrogen abstraction from 2,2-diphenyldipropane that match classical benchmarks, with performance evaluated across active spaces ranging up to (39e,39o).
What carries the argument
Entanglement forging that maps each qubit to a spatial orbital, thereby halving the qubit requirement, together with sample-based quantum diagonalization that projects the Schrödinger equation into a subspace of configurations sampled from the quantum device.
Load-bearing premise
The configurations sampled from the quantum device form a subspace representative enough to yield accurate projections of the Schrödinger equation for the activation and reaction energies, even at the largest active-space sizes tested.
What would settle it
A high-accuracy classical calculation such as density-matrix renormalization group or selected configuration interaction performed on the (39e,39o) active space that produces activation energies differing by more than chemical accuracy from the EF-SQD values would show the sampled subspace is insufficient.
Figures






read the original abstract
The simulation of electronic systems is an anticipated application for quantum-centric computers, i.e. heterogeneous architectures where classical and quantum processing units operate in concert. An important application is the computation of radical chain reactions, including those responsible for the photodegradation of composite materials used in aerospace engineering. Here, we compute the activation energy and reaction energy for hydrogen abstraction from 2,2-diphenyldipropane, used as a minimal model for a step in a radical chain reaction. Calculations are performed using a superconducting quantum processor of the IBM Heron family and classical computing resources. To this end, we combine a qubit-reduction technique called entanglement forging (EF) with sample-based quantum diagonalization (SQD), a method that projects the Schr\"{o}dinger equation into a subspace of configurations sampled from a quantum device. In conventional quantum simulations, a qubit represents a spin-orbital. In contrast, EF maps a qubit to a spatial orbital, reducing the required number of qubits by half. We provide a complete derivation and a detailed description of the combined EF and SQD approach, and we assess its accuracy across active spaces of varying sizes upto (39e,39o).
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript presents a quantum-centric simulation combining entanglement forging (EF) with sample-based quantum diagonalization (SQD) to compute activation and reaction energies for hydrogen abstraction from 2,2-diphenyldipropane on an IBM Heron superconducting processor. It supplies a full derivation of the EF-SQD workflow and reports accuracy assessments for active spaces ranging up to (39e,39o).
Significance. If the projection step is robust, the work demonstrates a viable route to larger active-space simulations with halved qubit count via EF, directly relevant to modeling radical reactions in aerospace composites. The explicit derivation and cross-size accuracy checks are positive features that would strengthen the contribution.
major comments (1)
- [Results and accuracy assessment sections (around the (39e,39o) calculations)] The central accuracy claim for the (39e,39o) space rests on the assumption that the SQD-sampled configurations, after EF mapping, form a representative subspace for projecting the Schrödinger equation. The configuration space dimension is binomial(78,39) ~ 10^22; the manuscript does not report independent metrics such as subspace overlap with a reference wavefunction or convergence of energies versus shot count or sample size, which is load-bearing for the reported activation/reaction energies.
minor comments (2)
- [Derivation of combined EF-SQD approach] Clarify in the methods whether the EF ansatz introduces any additional variational parameters beyond the standard SQD sampling procedure.
- [Tables and figures reporting numerical results] Add explicit error bars or shot-noise estimates to all tabulated energies and activation barriers.
Simulated Author's Rebuttal
We thank the referee for their careful reading of the manuscript and for the constructive feedback. We address the major comment below, providing additional context from our calculations and indicating the revisions we will make to strengthen the presentation of the accuracy assessment.
read point-by-point responses
-
Referee: [Results and accuracy assessment sections (around the (39e,39o) calculations)] The central accuracy claim for the (39e,39o) space rests on the assumption that the SQD-sampled configurations, after EF mapping, form a representative subspace for projecting the Schrödinger equation. The configuration space dimension is binomial(78,39) ~ 10^22; the manuscript does not report independent metrics such as subspace overlap with a reference wavefunction or convergence of energies versus shot count or sample size, which is load-bearing for the reported activation/reaction energies.
Authors: We agree that explicit validation of the sampled subspace is important for large active spaces. For active spaces up to (20e,20o) we directly compare SQD-EF energies to classical FCI or selected CI references, as shown in the accuracy assessment section; these comparisons confirm that the sampled configurations yield energies within chemical accuracy once the sample size exceeds a few thousand. For the (39e,39o) space, exact classical references are unavailable. However, we have conducted convergence tests with respect to both the number of sampled configurations and the number of shots per circuit; these tests appear in the supplementary information and show that the activation and reaction energies stabilize to within 1 kcal/mol once the sample size reaches approximately 10,000 configurations and 10^5 shots. We will revise the main text to explicitly reference these supplementary convergence plots and to add a short discussion of the statistical error bars and the rationale for expecting the sampled subspace to be representative at the reported sample sizes. Subspace overlap with a reference wavefunction cannot be provided for the (39e,39o) case because no classical exact solution exists. revision: partial
- Direct computation of subspace overlap with an exact reference wavefunction for the (39e,39o) active space, which is intractable classically.
Circularity Check
Minor self-citation on SQD/EF components; central projection remains independent
full rationale
The paper derives the combined EF-SQD method in detail and computes activation/reaction energies by projecting the Schrödinger equation onto a sampled subspace. No step reduces a claimed prediction to a fitted parameter by construction, nor does any load-bearing uniqueness theorem collapse to an unverified self-citation. Accuracy is assessed across active-space sizes with external classical benchmarks implied for smaller cases; hardware sampling introduces practical dependence but does not create definitional circularity. This yields a low score consistent with normal self-citation of prior method papers.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
for each sampled computational basis state with the wrong spin-resolved particle number,x k ∈χ, we compute the deviations ∆ k pσ =|(x k)pσ −n pσ| between the entries ofx k and those of the occupa- tion number distribution, and use them to define a probability distribution over the set of spin-orbitals pσ
-
[2]
we randomly flip a fixed number of 1s or 0s de- fined by the difference of the hamming weight of xk and desired number of electrons, according to the probability distribution in point 1, until the spin-resolved particle numbers assume target val- ues, thereby producing a new set of recovered con- figurationsχ R
-
[3]
we sampleKsubsets (batches) fromχ R, that we callχ b withb= 1. . . K. Each batch yields a sub- spaceS b of dimensiond, in which we project the Hamiltonian as in Eq. (1), producing an approxi- mationE b,|ψ b⟩to the ground eigenpair
-
[4]
we use the lowest energy across batches, minb Eb, as the best approximation to the ground-state energy, and we use the states|ψ b⟩to update the occupation number distribution, npσ = 1 K KX b=1 ⟨ψb|ˆa† pσˆapσ|ψb⟩.(2)
-
[5]
we repeat points 1-4 until convergence of the energy minb Eb and the occupation number distribution. In the first iteration of configuration recovery, instead of points 1-2, we postselect [32] computational basis states inχbased on particle number. In the current formu- lation of SQD, total spin conservation can only be ap- proximately enforced by adding ...
-
[6]
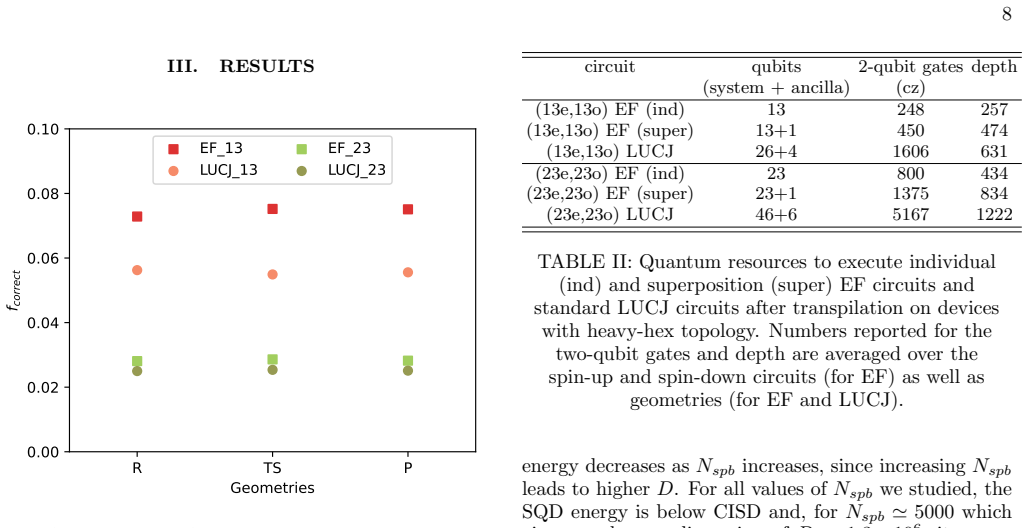
Quantum circuits Implementing the EF wavefunction in Eq. (18) requires constructing quantum circuits to (i) prepare a Slater de- terminant, e.g.|d µα⟩, or a superposition of Slater deter- minants, e.g. |dp µνα⟩= |dµα⟩+i p|dνα⟩ U p µν ,(19) withU p µν as in Eq. (6) and (ii) apply a uCCD operator. These circuits are shown in Fig. 2a, and described in detail...
-
[7]
Classical pre-processing We compute the activation energy ∆E ‡ =E TS −E R and the reaction energy ∆E=E P −E R in two differ- ent active spaces of sizes (13e,13o) and (23e,23o). These active spaces are spanned by intrinsic bond orbitals [67] located around CH • 3 and CH 4 where hydrogen abstrac- tion occurs for the active space (13e,13o) and around additio...
-
[8]
Details of quantum simulations We constructed the EF quantum circuits using the open- source ffsim library [54] and an in-house code to sup- ply the circuit parameters based on classical CCSD and ResHF calculations. We executed these quantum cir- cuits on IBM’s 133-qubit Heron superconducting quan- tum processoribm torino, gathering 500000 measurement out...
-
[9]
we choose a number of determinantsN det,
-
[10]
(17) defining Slater determinants|Φ(y µ)⟩,
we sampleN det auxiliary fields{y µ}Ndet µ=1 from the normal distribution in Eq. (17) defining Slater determinants|Φ(y µ)⟩,
-
[11]
we variationally optimize P µ cµ|Φ(yµ)⟩with respect toc µ andy µ. More specifically, we define ˜c µ(y) as the lowest-energy solu- tion of P µ⟨Φ(yν)| ˆH|Φ(y µ)⟩˜cµ(y) =E 0(y)P µ⟨Φ(yν)|Φ(yµ)⟩˜cµ(y) and minimizeE 0(y) as a function ofy. We use the resulting linear combination of non-orthogonal determinants as the initial point of the variational optimiza- ti...
-
[12]
Mangalgiri, Composite materials for aerospace appli- cations, Bull
P. Mangalgiri, Composite materials for aerospace appli- cations, Bull. Mater. Sci22, 657 (1999)
work page 1999
-
[13]
K. B. Mahat, I. Alarifi, A. Alharbi, and R. Asmatulu, Ef- fects of UV light on mechanical properties of carbon fiber reinforced pps thermoplastic composites, inMacromolec- ular Symposia, Vol. 365 (Wiley Online Library, 2016) pp. 157–168
work page 2016
-
[14]
D. Jung, Y. Mizutani, A. Todoroki, and W. Na, Effect of ultraviolet irradiation on the material properties and acoustic emission of a fiber-reinforced composite, Fibers and Polymers22, 1940 (2021)
work page 1940
-
[15]
T. S. Gates,On the use of accelerated test methods for characterization of advanced composite materials(Na- tional Aeronautics and Space Administration, Langley Research Center, 2003)
work page 2003
- [16]
-
[17]
U. Schulz, Accelerated testing: Nature and artificial weathering in the coatings industry, vincentz network gmbh & co, KG, Hannover, Germany (2009)
work page 2009
- [18]
-
[19]
B. Parveez, M. Kittur, I. A. Badruddin, S. Kamangar, M. Hussien, and M. Umarfarooq, Scientific advancements in composite materials for aircraft applications: a review, Polymers14, 5007 (2022)
work page 2022
-
[20]
Y. Zhu, K. Cao, M. Chen, and L. Wu, Synthesis of uv- responsive self-healing microcapsules and their potential application in aerospace coatings, ACS applied materials & interfaces11, 33314 (2019)
work page 2019
-
[21]
S. M. Arnold, D. Cebon, and M. Ashby,Materials selec- tion for aerospace systems, Tech. Rep. (2012)
work page 2012
-
[22]
K. Jayakrishna, V. R. Kar, M. T. Sultan, and M. Ra- jesh, Materials selection for aerospace components, in Sustainable composites for aerospace applications(Else- vier, 2018) pp. 1–18
work page 2018
-
[23]
D. B. Miracle, S. L. Donaldson, S. D. Henry, C. Moos- brugger, G. J. Anton, B. R. Sanders, N. Hrivnak, C. Ter- man, J. Kinson, K. Muldoon,et al.,ASM handbook, Vol. 21 (ASM international Materials Park, OH, 2001)
work page 2001
-
[24]
Quilter, Composites in aerospace applications, IHS White Paper444, 264 (2001)
A. Quilter, Composites in aerospace applications, IHS White Paper444, 264 (2001)
work page 2001
-
[25]
T. Lu, E. Solis-Ramos, Y. Yi, and M. Kumosa, Uv degra- dation model for polymers and polymer matrix compos- ites, Polym. Degrad. Stab154, 203 (2018)
work page 2018
-
[26]
S. Kiil, Model-based analysis of photoinitiated coating degradation under artificial exposure conditions, J. Coat. Technol. Res9, 375 (2012)
work page 2012
-
[27]
M. Evans, A statistical degradation model for the service life prediction of aircraft coatings: With a comparison to an existing methodology, Polymer Testing31, 46 (2012)
work page 2012
-
[28]
X. Meng, G. Jin, and R. Yang, A quantum chemical and molecular dynamics simulation study on photo-oxidative aging of polyethylene: Mechanism and differences be- tween crystalline and amorphous phases, Polym. Degrad. Stab217, 110536 (2023)
work page 2023
-
[29]
T. P. Gujarati, M. Motta, T. N. Friedhoff, J. E. Rice, N. Nguyen, P. K. Barkoutsos, R. J. Thompson, T. Smith, M. Kagele, M. Brei,et al., Quantum computation of re- actions on surfaces using local embedding, npj Quantum Inf9, 88 (2023)
work page 2023
-
[30]
J. L. Smialek, N. S. Jacobson,et al., Oxidation of high- temperature aerospace materials, High temperature ma- terials and mechanisms , 95 (2014)
work page 2014
-
[31]
Z. Shi, C. Zou, F. Zhou, and J. Zhao, Analysis of the mechanical properties and damage mechanism of carbon fiber/epoxy composites under uv aging, Materials15, 2919 (2022)
work page 2022
-
[32]
R. de Borst, Challenges in computational materials sci- ence: Multiple scales, multi-physics and evolving discon- tinuities, Computational Materials Science43, 1 (2008), proceedings of the 16th International Workshop on Com- putational Mechanics of Materials
work page 2008
-
[33]
N. Sai, K. Leung, J. Z´ ador, and G. Henkelman, First prin- ciples study of photo-oxidation degradation mechanisms in P3HT for organic solar cells, Phys. Chem. Chem. Phys 16, 8092 (2014)
work page 2014
- [34]
-
[35]
Y. Alexeev, M. Amsler, M. A. Barroca, S. Bassini, T. Battelle, D. Camps, D. Casanova, Y. J. Choi, F. T. Chong, C. Chung,et al., Quantum-centric supercomput- ing for materials science: A perspective on challenges and future directions, Fut. Gen. Comput. Sys160, 666 (2024)
work page 2024
- [36]
-
[37]
J. Robledo-Moreno, M. Motta, H. Haas, A. Javadi- Abhari, P. Jurcevic, W. Kirby, S. Martiel, K. Sharma, S. Sharma, T. Shirakawa,et al., Chemistry beyond exact solutions on a quantum-centric supercomputer, arXiv:2405.05068 (2024)
-
[38]
D. Kaliakin, A. Shajan, J. R. Moreno, Z. Li, A. Mi- tra, M. Motta, C. Johnson, A. A. Saki, S. Das, I. Sit- dikov,et al., Accurate quantum-centric simulations of supramolecular interactions, arXiv:2410.09209 (2024)
-
[39]
I. Liepuoniute, K. D. Doney, J. Robledo-Moreno, J. A. Job, W. S. Friend, and G. O. Jones, Quantum- centric study of methylene singlet and triplet states, arXiv:2411.04827 (2024)
-
[40]
S. Barison, J. R. Moreno, and M. Motta, Quantum- centric computation of molecular excited states with ex- tended sample-based quantum diagonalization, Quant. Sci. Tech10, 025034 (2025)
work page 2025
- [41]
- [42]
-
[43]
W. J. Huggins, J. R. McClean, N. C. Rubin, Z. Jiang, N. Wiebe, K. B. Whaley, and R. Babbush, Efficient and noise resilient measurements for quantum chemistry on near-term quantum computers, npj Quantum Inf7, 23 (2021)
work page 2021
- [44]
- [45]
-
[46]
J. A. Smolin, J. M. Gambetta, and G. Smith, Efficient method for computing the maximum-likelihood quantum state from measurements with additive Gaussian noise, Phys. Rev. Lett108, 070502 (2012)
work page 2012
-
[47]
I. Liepuoniute, M. Motta, T. Pellegrini, J. E. Rice, T. P. Gujarati, S. Gil, and G. O. Jones, Simulation of a Diels– Alder reaction on a quantum computer, Phys. Chem. Chem. Phys26, 25181 (2024)
work page 2024
- [48]
-
[49]
M. A. Castellanos, M. Motta, and J. E. Rice, Quantum computation ofπ→π* and n→π* excited states of aro- matic heterocycles, Mol. Phys122, e2282736 (2024)
work page 2024
-
[50]
H. F. Trotter, On the product of semi-groups of opera- tors, Proc. Am. Math. Soc10, 545 (1959)
work page 1959
-
[51]
Hubbard, Calculation of partition functions, Phys
J. Hubbard, Calculation of partition functions, Phys. Rev. Lett3, 77 (1959)
work page 1959
-
[52]
R. L. Stratonovich, On a method of calculating quantum distribution functions, Sov. Phys. Doklady2, 416 (1957)
work page 1957
-
[53]
S. Zhang and H. Krakauer, Quantum monte carlo method using phase-free random walks with slater determinants, Phys. Rev. Lett90, 136401 (2003)
work page 2003
-
[54]
M. Motta and S. Zhang, Ab initio computations of molec- ular systems by the auxiliary-field quantum monte carlo method, WIREs Comput. Mol. Sci8, e1364 (2018)
work page 2018
-
[55]
D. J. Thouless, Stability conditions and nuclear rotations in the hartree-fock theory, Nuc. Phys21, 225 (1960)
work page 1960
-
[56]
R. Balian and E. Brezin, Nonunitary Bogoliubov trans- formations and extension of Wick’s theorem, Il Nuovo Cimento B64, 37 (1969)
work page 1969
-
[57]
Bremond, Hartree-Fock methods for degenerate cases, Nuc
B. Bremond, Hartree-Fock methods for degenerate cases, Nuc. Phys58, 687 (1964)
work page 1964
-
[58]
H. Fukutome, Theory of resonating quantum fluctuations in a fermion system: resonating Hartree-Fock approxima- tion, Progr. Theor. Phys80, 417 (1988)
work page 1988
-
[59]
P. ˚A. Malmqvist, Calculation of transition density matri- ces by nonunitary orbital transformations, Int. J. Quan- tum Chem30, 479 (1986)
work page 1986
-
[60]
A. J. Thom and M. Head-Gordon, Hartree–Fock solu- tions as a quasidiabatic basis for nonorthogonal configu- ration interaction, J. Chem. Phys131, 124113 (2009)
work page 2009
-
[61]
E. J. Sundstrom and M. Head-Gordon, Non-orthogonal configuration interaction for the calculation of multielec- tron excited states, J. Chem. Phys140, 114103 (2014)
work page 2014
-
[62]
M. Reck, A. Zeilinger, H. J. Bernstein, and P. Bertani, Experimental realization of any discrete unitary opera- tor, Phys. Rev. Lett73, 58 (1994)
work page 1994
-
[63]
W. R. Clements, P. C. Humphreys, B. J. Metcalf, W. S. Kolthammer, and I. A. Walmsley, Optimal design for uni- versal multiport interferometers, Optica3, 1460 (2016)
work page 2016
- [64]
-
[65]
The ffsim developers, ffsim: Faster simulations of fermionic quantum circuits
- [66]
-
[67]
Y. Matsuzawa and Y. Kurashige, Jastrow-type decom- position in quantum chemistry for low-depth quantum circuits, J. Chem. Theory Comput16, 944 (2020)
work page 2020
- [68]
-
[69]
Generalized swap networks for near-term quantum computing
B. O’Gorman, W. J. Huggins, E. G. Rieffel, and K. B. Whaley, Generalized swap networks for near-term quan- tum computing, arXiv:1905.05118 (2019)
work page internal anchor Pith review Pith/arXiv arXiv 1905
-
[70]
A. Rivaton, L. Moreau, and J.-L. Gardette, Photo- oxidation of phenoxy resins at long and short wave- lengths—ii. mechanisms of formation of photoproducts, Polym. Degrad. Stab58, 333 (1997)
work page 1997
-
[71]
E. Yousif and R. Haddad, Photodegradation and photostabilization of polymers, especially polystyrene, SpringerPlus2, 1 (2013). 14
work page 2013
-
[72]
B. Mailhot, S. Morlat-Th´ erias, P.-O. Bussi` ere, and J.-L. Gardette, Study of the degradation of an epoxy/amine resin, 2, Macromolecular Chemistry and Physics206, 585 (2005)
work page 2005
-
[73]
2,2-diphenylpropane (compound)
-
[74]
A. D. Becke, Density-functional thermochemistry. III. the role of exact exchange, J. Chem. Phys98, 5648 (1993)
work page 1993
- [75]
-
[76]
Dunning, Thom H., Gaussian basis sets for use in correlated molecular calculations
J. Dunning, Thom H., Gaussian basis sets for use in correlated molecular calculations. I. the atoms boron through neon and hydrogen, J. Chem. Phys90, 1007 (1989)
work page 1989
-
[77]
Y. Cao, T. Balduf, M. D. Beachy, M. C. Bennett, A. D. Bochevarov, A. Chien, P. A. Dub, K. G. Dyall, J. W. Furness, M. D. Halls, T. F. Hughes, L. D. Jacobson, H. S. Kwak, D. S. Levine, D. T. Mainz, I. Moore, Kevin B., M. Svensson, P. E. Videla, M. A. Watson, and R. A. Friesner, Quantum chemical package Jaguar: a survey of recent developments and unique fea...
work page 2024
-
[78]
G. Knizia, Intrinsic atomic orbitals: An unbiased bridge between quantum theory and chemical concepts, Journal of Chemical Theory and Computation9, 4834 (2013), pMID: 26583402, https://doi.org/10.1021/ct400687b
-
[79]
Q. Sun, T. C. Berkelbach, N. S. Blunt, G. H. Booth, S. Guo, Z. Li, J. Liu, J. D. McClain, E. R. Sayfutyarova, S. Sharma,et al., PySCF: the Python-based simulations of chemistry framework, WIREs Comput. Mol. Sci8, e1340 (2018)
work page 2018
-
[80]
Q. Sun, X. Zhang, S. Banerjee, P. Bao, M. Barbry, N. S. Blunt, N. A. Bogdanov, G. H. Booth, J. Chen, Z.-H. Cui,et al., Recent developments in the PySCF program package, J. Chem. Phys153, 024109 (2020)
work page 2020
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.