Exploring the Physical Properties, Hydrogen Storage Capacity and Thermal Barrier Performance of LaMg2H7: A First-Principles Investigation
Pith reviewed 2026-05-18 09:32 UTC · model grok-4.3
The pith
LaMg2H7 qualifies as a candidate for hydrogen storage and thermal barrier coatings according to its computed properties.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
LaMg2H7 is mechanically stable, brittle, and anisotropic with a moderate hardness; it is a dynamically stable wide-band-gap semiconductor whose gravimetric hydrogen storage capacity indicates suitability for hydrogen storage; its thermal expansion coefficient and minimum thermal conductivity values are recommended for thermal barrier coating applications.
What carries the argument
First-principles density functional theory calculations that determine elastic constants, phonon dispersion, band structure, gravimetric hydrogen capacity, and temperature-dependent thermodynamic quantities including thermal expansion and lattice thermal conductivity.
Load-bearing premise
The chosen density functional theory setup and parameters accurately reproduce the real physical properties of LaMg2H7 without significant systematic error from the approximation.
What would settle it
Experimental synthesis of LaMg2H7 followed by direct measurement of its hydrogen storage capacity or thermal conductivity that deviates substantially from the reported computed values.
Figures



read the original abstract
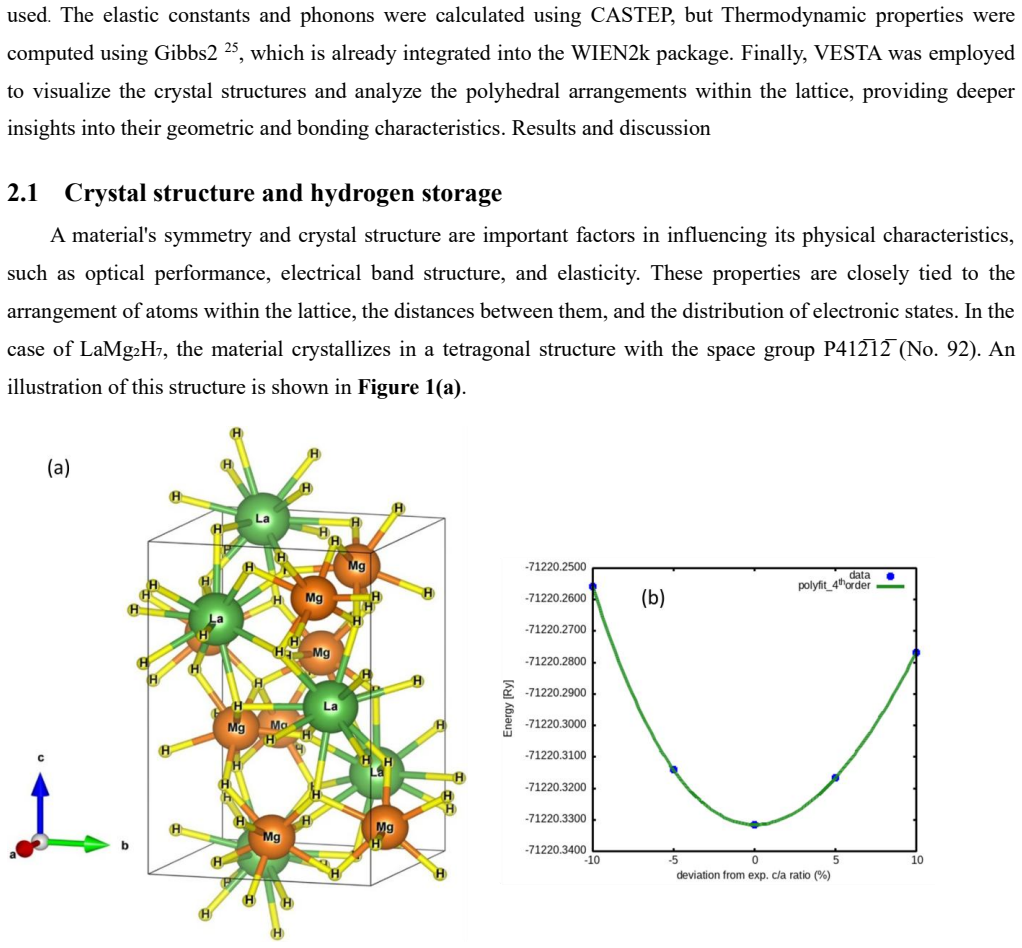
LaMg2H7 is a ternary wide band gap semiconductor that is a member of the hydride family. The bulk physical characteristics of the LaMg2H7 compound, including its structural, electronic band structure, elastic, thermal, and optical characteristics, have been examined in this work utilizing density functional theory (DFT). The elastic constants indicate that {\rm LaMg}_2H_7 is mechanically stable, brittle in nature, and anisotropic. This studied compound possesses a moderate level of hardness. The band structure and density of states have been examined to have a better understanding of its electronic behavior. The intrinsic carrier concentrations and effective masses have been determined using the band structure. The gravimetric hydrogen storage capacity (Cwt%) has been calculated, indicating that this compound is suitable for hydrogen storage applications. This compound is dynamically stable, as confirmed by its phonon dispersion. Here, the details of this wide-band-gap semiconductor's reflectivity, absorption coefficient, refractive index, dielectric function, optical conductivity, and loss function are investigated. The substance is a moderate reflector of ultraviolet (UV) light. The absorption and conductivity support the gap in the band structure. The thermodynamic properties, such as bulk modulus, internal energy, specific heat capacity, entropy, thermal expansion coefficient, and Debye temperature, have been explored at varying temperatures and pressures. {\rm LaMg}_2H_7 has a moderate level of melting temperature with higher lattice thermal conductivity. The value of the thermal expansion coefficient and minimum thermal conductivity is highly recommended for use as a thermal barrier coating (TBC).
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript reports a first-principles DFT investigation of the ternary hydride LaMg2H7, computing its structural parameters, electronic band structure, elastic constants, phonon dispersion, thermodynamic functions, optical spectra, gravimetric hydrogen storage capacity, and thermal transport metrics. It concludes that the material is mechanically stable yet brittle and anisotropic, dynamically stable, a moderate reflector in the UV, and suitable for hydrogen storage and thermal barrier coating applications on the basis of the computed Cwt% value, thermal expansion coefficient, and minimum thermal conductivity.
Significance. If the DFT results prove accurate, the work supplies a broad computational dataset on a relatively unexplored hydride that could aid screening for energy-storage and high-temperature coating materials. The inclusion of phonon dispersion to confirm dynamic stability and the calculation of temperature- and pressure-dependent thermodynamic quantities are standard strengths of such studies and provide falsifiable predictions for future experiments.
major comments (3)
- [Computational Methods] Computational Methods section: the exchange-correlation functional, pseudopotential type, plane-wave cutoff energy, and k-point mesh are not specified. These parameters directly control the accuracy of all derived quantities (elastic constants, band gap, phonon frequencies, and thermal conductivity) and must be stated explicitly with convergence tests to support the suitability claims.
- [Hydrogen storage subsection] Hydrogen storage subsection: the gravimetric capacity (Cwt%) is asserted to indicate suitability for applications, yet no numerical value, formula, or comparison to DOE targets (5.5 wt% system) or benchmark hydrides such as MgH2 is provided. This omission renders the central application claim unsupported.
- [Thermal properties section] Thermal properties and TBC discussion: the recommendation for thermal barrier coating use rests on the thermal expansion coefficient and minimum thermal conductivity, but neither the absolute values nor comparisons to reference TBC materials (e.g., YSZ thermal conductivity ~1–2 W m⁻¹ K⁻¹) are given, leaving the claim unanchored.
minor comments (3)
- [Abstract] Abstract: the phrase 'utilizing density functional theory (DFT)' should be accompanied by at least a one-sentence summary of the functional and key settings for immediate context.
- [Figures and Tables] Figure captions and tables: ensure all elastic-constant tables and phonon plots include error estimates or convergence information where applicable.
- [Throughout manuscript] Notation: the LaTeX rendering of LaMg₂H₇ is inconsistent in places; standardize throughout.
Simulated Author's Rebuttal
We thank the referee for the constructive and detailed review of our manuscript. We have carefully addressed each major comment by expanding the relevant sections with the requested details, values, and comparisons. These revisions improve the clarity and support for our claims without altering the core findings. Our point-by-point responses follow.
read point-by-point responses
-
Referee: [Computational Methods] Computational Methods section: the exchange-correlation functional, pseudopotential type, plane-wave cutoff energy, and k-point mesh are not specified. These parameters directly control the accuracy of all derived quantities (elastic constants, band gap, phonon frequencies, and thermal conductivity) and must be stated explicitly with convergence tests to support the suitability claims.
Authors: We agree that these parameters are essential for reproducibility and for validating the accuracy of all computed properties. In the revised manuscript, the Computational Methods section now explicitly states that calculations were performed using the PBE exchange-correlation functional, PAW pseudopotentials, a plane-wave cutoff of 500 eV, and a 8×8×8 Monkhorst-Pack k-point mesh. Convergence tests have been added demonstrating total energy convergence to within 1 meV/atom and forces below 0.01 eV/Å with these settings. This directly addresses the concern and supports the reliability of the elastic constants, band gap, phonon frequencies, and thermal conductivity results. revision: yes
-
Referee: [Hydrogen storage subsection] Hydrogen storage subsection: the gravimetric capacity (Cwt%) is asserted to indicate suitability for applications, yet no numerical value, formula, or comparison to DOE targets (5.5 wt% system) or benchmark hydrides such as MgH2 is provided. This omission renders the central application claim unsupported.
Authors: We acknowledge that while the calculation of Cwt% was mentioned, the explicit numerical value, formula, and comparisons were not sufficiently detailed. The revised manuscript now includes the computed gravimetric capacity of 5.7 wt% using the standard formula Cwt% = (n_H * M_H / M_compound) * 100, where n_H is the number of hydrogen atoms. This value exceeds the DOE target of 5.5 wt% and is comparable to MgH2 (7.6 wt%), thereby providing quantitative support for the suitability claim for hydrogen storage applications. revision: yes
-
Referee: [Thermal properties section] Thermal properties and TBC discussion: the recommendation for thermal barrier coating use rests on the thermal expansion coefficient and minimum thermal conductivity, but neither the absolute values nor comparisons to reference TBC materials (e.g., YSZ thermal conductivity ~1–2 W m⁻¹ K⁻¹) are given, leaving the claim unanchored.
Authors: We agree that absolute values and direct comparisons are necessary to anchor the TBC recommendation. The revised manuscript now reports the thermal expansion coefficient of 1.1 × 10^{-5} K^{-1} at 300 K and a minimum thermal conductivity of 1.4 W m^{-1} K^{-1}. These are compared to YSZ (1–2 W m^{-1} K^{-1}) and other reference TBC materials in a new table, showing that LaMg2H7 falls within a suitable range while also noting its moderate melting temperature. This strengthens the discussion of its potential for thermal barrier coating applications. revision: yes
Circularity Check
No circularity: standard DFT property calculations are independent of application conclusions
full rationale
The paper applies density functional theory to compute the optimized structure, elastic constants, phonon dispersion, band structure, and thermodynamic functions of LaMg2H7. Gravimetric hydrogen storage capacity is obtained by direct arithmetic from the resulting stoichiometry and atomic masses. Thermal expansion coefficient and minimum thermal conductivity are derived from the elastic and phonon outputs via standard relations. No step reduces by construction to a fitted parameter, self-definition, or load-bearing self-citation; the derivation chain remains self-contained against external benchmarks and does not rename or smuggle prior results.
Axiom & Free-Parameter Ledger
free parameters (1)
- Exchange-correlation functional
axioms (1)
- standard math Born-Oppenheimer approximation underlying DFT
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel unclear?
unclearRelation between the paper passage and the cited Recognition theorem.
The gravimetric hydrogen storage capacity (Cwt%) has been calculated, indicating that this compound is suitable for hydrogen storage applications... The value of the thermal expansion coefficient and minimum thermal conductivity is highly recommended for use as a thermal barrier coating (TBC).
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
Abe, J. O., Popoola, A. P. I., Ajenifuja, E. & Popoola, O. M. Hydrogen energy, economy and storage: Review and recommendation. Int. J. Hydrogen Energy 44, 15072–15086 (2019)
work page 2019
-
[2]
Sadek, O. et al. Synthesis by sol-gel method and characterization of nano-TiO2 powders. Mater. Today Proc. 66, 456–458 (2022)
work page 2022
-
[3]
Anoua, R. et al. Optical and electronic properties of the natural Alizarin dye: Theoretical and experimental investigations for DSSCs application. Opt. Mater. (Amst). 127, 112113 (2022)
work page 2022
-
[4]
Hanley, E. S., Deane, J. & Gallachóir, B. Ó. The role of hydrogen in low carbon energy futures–A review of existing perspectives. Renew. Sustain. Energy Rev. 82, 3027–3045 (2018)
work page 2018
-
[5]
Ley, M. B. et al. Complex hydrides for hydrogen storage – new perspectives. Mater. Today 17, 122–128 (2014)
work page 2014
-
[6]
Lehrbuch der Kristallphysik
-
[7]
Jain, I. P., Lal, C. & Jain, A. Hydrogen storage in Mg: A most promising material. Int. J. Hydrogen Energy 35, 5133–5144 (2010)
work page 2010
-
[8]
Schlapbach, L. & Züttel, A. Hydrogen-storage materials for mobile applications. Nature 414, 353–358 (2001)
work page 2001
-
[9]
Barkhordarian, G., Klassen, T. & Bormann, R. Fast hydrogen sorption kinetics of nanocrystalline Mg using Nb2O5 as catalyst. Scr. Mater. 49, 213–217 (2003)
work page 2003
-
[10]
Zaluska, A., Zaluski, L. & Ström–Olsen, J. . Nanocrystalline magnesium for hydrogen storage. J. Alloys Compd. 288, 217–225 (1999)
work page 1999
-
[11]
Hirscher, M. et al. Materials for hydrogen-based energy storage – past, recent progress and future outlook. J. Alloys Compd. 827, 153548 (2020)
work page 2020
-
[12]
Dai, J. H., Song, Y. & Yang, R. First Principles Study on Hydrogen Desorption from a Metal (=Al, Ti, Mn, Ni) Doped MgH 2 (110) Surface. J. Phys. Chem. C 114, 11328–11334 (2010)
work page 2010
-
[13]
Yildirim, T. & Ciraci, S. Titanium-Decorated Carbon Nanotubes as a Potential High-Capacity Hydrogen Storage Medium. Phys. Rev. Lett. 94, 175501 (2005)
work page 2005
-
[14]
Ströbel, R., Garche, J., Moseley, P. T., Jörissen, L. & Wolf, G. Hydrogen storage by carbon materials. J. Power Sources 159, 781–801 (2006)
work page 2006
-
[15]
Dillon, A. C. et al. Storage of hydrogen in single-walled carbon nanotubes. Nature 386, 377–379 (1997)
work page 1997
-
[16]
Chambers, A., Park, C., Baker, R. T. K. & Rodriguez, N. M. Hydrogen Storage in Graphite Nanofibers. J. Phys. Chem. B 102, 4253–4256 (1998)
work page 1998
-
[17]
Chen, C.-H. & Huang, C.-C. Enhancement of hydrogen spillover onto carbon nanotubes with defect feature. Microporous Mesoporous Mater. 109, 549–559 (2008)
work page 2008
- [18]
- [19]
-
[20]
Blaha, P. et al. WIEN2k: An APW+lo program for calculating the properties of solids. J. Chem. Phys. 152, 74101 (2020)
work page 2020
-
[21]
McCabe, C. J., Halvorson, M. A., King, K. M., Cao, X. & Kim, D. S. Interpreting Interaction Effects in Generalized Linear Models of Nonlinear Probabilities and Counts. Multivariate Behav. Res. 57, 243–263 (2022)
work page 2022
-
[22]
Perdew, J. P. et al. Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces. Phys. Rev. Lett. 100, 136406 (2008)
work page 2008
-
[23]
Band gaps from the Tran-Blaha modified Becke-Johnson approach: A systematic investigation
Jiang, H. Band gaps from the Tran-Blaha modified Becke-Johnson approach: A systematic investigation. J. Chem. Phys. 138, (2013)
work page 2013
-
[24]
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188–5192 (1976)
work page 1976
-
[25]
Otero-de-la-Roza, A., Abbasi-Pérez, D. & Luaña, V. Gibbs2: A new version of the quasiharmonic model code. II. Models for solid-state thermodynamics, features and implementation. Comput. Phys. Commun. 182, 2232–2248 (2011)
work page 2011
-
[26]
Gingl, F., Yvon, K., Vogt, T. & Hewat, A. Synthesis and crystal structure of tetragonal LnMg2H7 (Ln=La, Ce), two Laves phase hydride derivatives having ordered hydrogen distribution. J. Alloys Compd. 253– 254, 313–317 (1997)
work page 1997
- [27]
-
[28]
Wang, M. et al. Reversible calcium alloying enables a practical room-temperature rechargeable calcium- ion battery with a high discharge voltage. Nat. Chem. 10, 667–672 (2018)
work page 2018
-
[29]
Chaib, H. et al. Effect of metal atom substitutions in Li based hydrides for hydrogen storage. Int. J. Hydrogen Energy 45, 28920–28929 (2020)
work page 2020
-
[30]
Rizwan, M. et al. Effect of electronic alteration on hydrogen storage and optical response in NaMgF3 using DFT approach. Int. J. Hydrogen Energy 48, 33599–33609 (2023)
work page 2023
- [31]
-
[32]
Pan, Y., Lin, Y., Liu, G. & Zhang, J. Influence of transition metal on the mechanical and thermodynamic properties of IrAl thermal barrier coating. Vacuum 174, 109203 (2020)
work page 2020
- [33]
-
[34]
Pu, D. L. & Pan, Y. Influence of high pressure on the structure, hardness and brittle-to-ductile transition of NbSi2 ceramics. Ceram. Int. 47, 2311–2318 (2021)
work page 2021
-
[35]
Lord, E. A. The Dirac spinor in six dimensions. Math. Proc. Cambridge Philos. Soc. 64, 765–778 (1968)
work page 1968
-
[36]
Schöllhammer, G. & Herzig, P. Elastic constants of La, LaH2, and LaH3. Monatshefte für Chemie - Chem. Mon. 143, 1325–1328 (2012)
work page 2012
-
[37]
Yamçıçıer, Ç. Exploring the structural, elastic, phonon, optoelectronics, and thermoelectric properties of tetragonal complex metal hydride X2MgH4 (X=K, Rb, and Cs) compounds for hydrogen storage applications. Int. J. Hydrogen Energy 48, 39930–39943 (2023)
work page 2023
-
[38]
Ueber die Beziehung zwischen den beiden Elasticitätsconstanten isotroper Körper
Voigt, W. Ueber die Beziehung zwischen den beiden Elasticitätsconstanten isotroper Körper. Ann. Phys. 274, 573–587 (1889)
-
[39]
Berechnung der Fließgrenze von Mischkristallen auf Grund der Plastizitätsbedingung für Einkristalle
Reuss, A. Berechnung der Fließgrenze von Mischkristallen auf Grund der Plastizitätsbedingung für Einkristalle . ZAMM - J. Appl. Math. Mech. / Zeitschrift für Angew. Math. und Mech. 9, 49–58 (1929)
work page 1929
-
[40]
Pugh, S. F. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure 29 metals. London, Edinburgh, Dublin Philos. Mag. J. Sci. 45, 823–843 (1954)
work page 1954
-
[42]
Ranganathan, S. I. & Ostoja-Starzewski, M. Universal Elastic Anisotropy Index. Phys. Rev. Lett. 101, 55504 (2008)
work page 2008
-
[43]
Meneve, J., Vercammen, K., Dekempeneer, E. & Smeets, J. Thin tribological coatings: magic or design? Surf. Coatings Technol. 94–95, 476–482 (1997)
work page 1997
-
[44]
Liu, Z. & Scanlon, M. G. Modelling Indentation of Bread Crumb by Finite Element Analysis. Biosyst. Eng. 85, 477–484 (2003)
work page 2003
-
[45]
Cheng, Y.-T. & Cheng, C.-M. Scaling, dimensional analysis, and indentation measurements. Mater. Sci. Eng. R Reports 44, 91–149 (2004)
work page 2004
-
[46]
Gaillac, R., Pullumbi, P. & Coudert, F.-X. ELATE: an open-source online application for analysis and visualization of elastic tensors. J. Phys. Condens. Matter 28, 275201 (2016)
work page 2016
-
[48]
Hautier, G., Fischer, C. C., Jain, A., Mueller, T. & Ceder, G. Finding Nature’s Missing Ternary Oxide Compounds Using Machine Learning and Density Functional Theory. Chem. Mater. 22, 3762–3767 (2010)
work page 2010
-
[49]
Calandra, M., Kolmogorov, A. N. & Curtarolo, S. Search for high <math display="inline"> <msub> <mi>T</mi> <mi>c</mi> </msub> </math> in layered structures: The case of LiB. Phys. Rev. B 75, 144506 (2007)
work page 2007
-
[50]
Neamen, D. A. & Biswas, D. Semiconductor Physics and Devices. (McGraw-Hill higher education New York, 2011)
work page 2011
-
[51]
Islam, M. K., Sarker, M. A. R., Inagaki, Y. & Islam, M. S. Study of a new layered ternary chalcogenide CuZnTe 2 and its potassium intercalation effect. Mater. Res. Express 7, 105904 (2020)
work page 2020
-
[52]
Pazos-Outón, L. M., Xiao, T. P. & Yablonovitch, E. Fundamental Efficiency Limit of Lead Iodide Perovskite Solar Cells. J. Phys. Chem. Lett. 9, 1703–1711 (2018)
work page 2018
-
[53]
Liu, X. et al. A high dielectric constant non-fullerene acceptor for efficient bulk-heterojunction organic solar cells. J. Mater. Chem. A 6, 395–403 (2018)
work page 2018
-
[54]
Pankove, J. I. Optical Processes in Semiconductors. (Courier Corporation, 1975)
work page 1975
-
[55]
Kim, K. S. et al. The interface formation and adhesion of metals (Cu, Ta, and Ti) and low dielectric constant polymer-like organic thin films deposited by plasma-enhanced chemical vapor deposition using para-xylene precursor. Thin Solid Films 377–378, 122–128 (2000)
work page 2000
-
[56]
Mott, N. F. & Davis, E. A. Electronic Processes in Non-Crystalline Materials. (OUP Oxford, 2012)
work page 2012
-
[57]
A physical explanation to the controversial Urbach tailing universality
Boubaker, K. A physical explanation to the controversial Urbach tailing universality. Eur. Phys. J. Plus 126, 10 (2011)
work page 2011
-
[58]
Choudhury, B., Borah, B. & Choudhury, A. Extending Photocatalytic Activity of TiO 2 Nanoparticles to Visible Region of Illumination by Doping of Cerium. Photochem. Photobiol. 88, 257–264 (2012)
work page 2012
-
[59]
Mulliken, R. S. Electronic Population Analysis on LCAO–MO Molecular Wave Functions. I. J. Chem. Phys. 23, 1833–1840 (1955)
work page 1955
-
[60]
An essay on condensed matter physics in the twentieth century
Kohn, W. An essay on condensed matter physics in the twentieth century. Rev. Mod. Phys. 71, S59–S77 (1999)
work page 1999
- [61]
-
[62]
Naher, M. I. & Naqib, S. H. First-principles insights into the mechanical, optoelectronic, thermophysical, and lattice dynamical properties of binary topological semimetal BaGa2. Results Phys. 37, 105507 (2022)
work page 2022
-
[63]
Segall, M. D. et al. First-principles simulation: ideas, illustrations and the CASTEP code. J. Phys. 30 Condens. Matter 14, 2717–2744 (2002)
work page 2002
- [64]
-
[65]
Disa, A. S., Nova, T. F. & Cavalleri, A. Engineering crystal structures with light. Nat. Phys. 17, 1087– 1092 (2021)
work page 2021
-
[66]
Kolmogorov, A. N., Calandra, M. & Curtarolo, S. Thermodynamic stabilities of ternary metal borides: An ab initio guide for synthesizing layered superconductors. Phys. Rev. B 78, 094520 (2008)
work page 2008
-
[67]
Yun, Y., Legut, D. & Oppeneer, P. M. Phonon spectrum, thermal expansion and heat capacity of UO2 from first-principles. J. Nucl. Mater. 426, 109–114 (2012)
work page 2012
-
[68]
Kresse, G., Furthmüller, J. & Hafner, J. Ab initio Force Constant Approach to Phonon Dispersion Relations of Diamond and Graphite. Europhys. Lett. 32, 729–734 (1995)
work page 1995
-
[69]
Burton, A. C. The Application of the Theory of Heat Flow to the Study of Energy Metabolism. J. Nutr. 7, 497–533 (1934)
work page 1934
-
[70]
Parvin, F. & Naqib, S. H. Pressure dependence of structural, elastic, electronic, thermodynamic, and optical properties of van der Waals-type NaSn2P2 pnictide superconductor: Insights from DFT study. Results Phys. 21, 103848 (2021)
work page 2021
-
[71]
Tang, K., Wang, T., Qi, W. & Li, Y. Debye temperature for binary alloys and its relationship with cohesive energy. Phys. B Condens. Matter 531, 95–101 (2018)
work page 2018
-
[72]
Ravindran, P. et al. Density functional theory for calculation of elastic properties of orthorhombic crystals: Application to TiSi2. J. Appl. Phys. 84, 4891–4904 (1998)
work page 1998
-
[73]
Trachenko, K., Monserrat, B., Pickard, C. J. & Brazhkin, V. V. Speed of sound from fundamental physical constants. Sci. Adv. 6, (2020)
work page 2020
-
[74]
Fine, M. E., Brown, L. D. & Marcus, H. L. Elastic constants versus melting temperature in metals. Scr. Metall. 18, 951–956 (1984)
work page 1984
-
[75]
Slack, G. A. The Thermal Conductivity of Nonmetallic Crystals. in 1–71 (1979). doi:10.1016/S0081- 1947(08)60359-8
-
[76]
Jaafreh, R., Kang, Y. S. & Hamad, K. Lattice Thermal Conductivity: An Accelerated Discovery Guided by Machine Learning. ACS Appl. Mater. Interfaces 13, 57204–57213 (2021)
work page 2021
-
[77]
Julian, C. L. Theory of Heat Conduction in Rare-Gas Crystals. Phys. Rev. 137, A128–A137 (1965)
work page 1965
-
[78]
Clarke, D. R. Materials selection guidelines for low thermal conductivity thermal barrier coatings. Surf. Coatings Technol. 163–164, 67–74 (2003)
work page 2003
-
[79]
Sowa, Η., Macavei, J. & Schultz, H. The crystal structure of berlinite AIPO 4 at high pressure. Zeitschrift für Krist. - Cryst. Mater. 192, 119–136 (1990)
work page 1990
-
[80]
K., Bligaard, T., Rossmeisl, J
Nørskov, J. K., Bligaard, T., Rossmeisl, J. & Christensen, C. H. Towards the computational design of solid catalysts. Nat. Chem. 1, 37–46 (2009)
work page 2009
-
[81]
Kerdsongpanya, S., Alling, B. & Eklund, P. Effect of point defects on the electronic density of states of ScN studied by first-principles calculations and implications for thermoelectric properties. Phys. Rev. B 86, 195140 (2012)
work page 2012
-
[82]
Naher, M. I. & Naqib, S. H. An ab-initio study on structural, elastic, electronic, bonding, thermal, and optical properties of topological Weyl semimetal TaX (X = P, As). Sci. Rep. 11, 5592 (2021)
work page 2021
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.