A Unification of Discrete, Gaussian, and Simplicial Diffusion
Pith reviewed 2026-05-16 21:16 UTC · model grok-4.3
The pith
Discrete, Gaussian, and simplicial diffusion arise as different parameterizations and large-population limits of the Wright-Fisher population genetics model.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
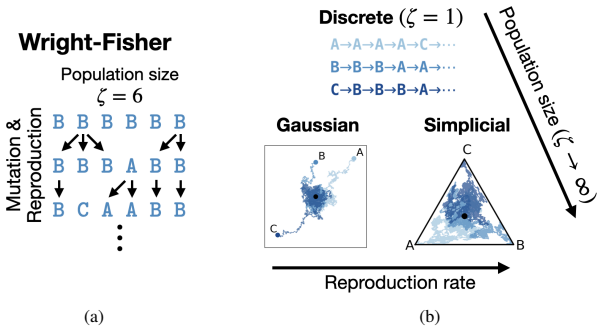
All three major methods of diffusion for discrete sequences—discrete diffusion, Gaussian diffusion in Euclidean space, and diffusion on the simplex—are different parameterizations of the Wright-Fisher population genetics model. Simplicial and Gaussian diffusion emerge as two large-population limits of this process. The resulting theory formally connects the likelihoods and hyperparameters across the three families and supplies stable stochastic processes for simplicial diffusion drawn from the genetics literature. A single trained model can then perform diffusion in any of the three domains at test time.
What carries the argument
The Wright-Fisher model, a finite-population stochastic process from population genetics that tracks changes in allele frequencies under drift; it supplies the common dynamics whose specific discretizations and scaling limits recover the three diffusion schemes.
If this is right
- Likelihoods and hyperparameters of discrete, Gaussian, and simplicial diffusion become formally interchangeable through their shared Wright-Fisher parameterization.
- Stable numerical schemes for simplicial diffusion follow directly from existing mathematical genetics results.
- A single model can be trained once and then deployed for diffusion in any of the three domains at test time.
- Wright-Fisher simplicial diffusion achieves higher stability and better performance than prior simplicial methods on conditional DNA generation tasks.
- Models trained jointly across domains remain competitive with models trained on any one domain separately.
Where Pith is reading between the lines
- Varying the effective population size parameter inside the Wright-Fisher framework could yield new families of diffusion schedules that interpolate continuously between the three regimes.
- The large-population limits may clarify when Gaussian approximations remain accurate for discrete data and when they break down.
- Tools developed for analyzing convergence rates in population-genetics models could be repurposed to study sampling efficiency and mixing times in diffusion generative models.
- The unification suggests testing whether other discrete generative processes outside diffusion, such as certain autoregressive or flow models, also admit Wright-Fisher interpretations.
Load-bearing premise
The specific discretizations and stochastic processes used in existing discrete, Gaussian, and simplicial diffusion models match instances or limits of Wright-Fisher dynamics exactly, without extra approximations that would break equivalence of likelihoods and hyperparameters.
What would settle it
A side-by-side computation of exact transition probabilities or marginal likelihoods between a Wright-Fisher-derived simplicial process and a standard simplex diffusion implementation that shows systematic, non-negligible differences persisting even after population-size scaling is accounted for.
Figures











read the original abstract
To model discrete sequences such as DNA, proteins, and language using diffusion, practitioners must choose between three major methods: diffusion in discrete space, Gaussian diffusion in Euclidean space, or diffusion on the simplex. Despite their shared goal, these models have disparate algorithms, theoretical structures, and tradeoffs: discrete diffusion has the most natural domain, Gaussian diffusion has more mature algorithms, and diffusion on the simplex in principle combines the strengths of the other two but in practice suffers from a numerically unstable stochastic processes. Ideally we could see each of these models as instances of the same underlying framework, and enable practitioners to switch between models for downstream applications. However previous theories have only considered connections in special cases. Here we build a theory unifying all three methods of discrete diffusion as different parameterizations of the same underlying process: the Wright-Fisher population genetics model. In particular, we find simplicial and Gaussian diffusion as two large-population limits. Our theory formally connects the likelihoods and hyperparameters of these models and leverages decades of mathematical genetics literature to unlock stable simplicial diffusion. Finally, we relieve the practitioner of balancing model trade-offs by demonstrating it is possible to train a single model that can perform diffusion in any of these three domains at test time. Our experiments show that Wright-Fisher simplicial diffusion is more stable and outperforms previous simplicial diffusion models on conditional DNA generation. We also show that we can train models on multiple domains at once that are competitive with models trained on any individual domain.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript claims to unify discrete, Gaussian, and simplicial diffusion models for discrete sequences by framing them as different parameterizations of the Wright-Fisher population genetics model. Simplicial and Gaussian diffusion are derived as two large-population limits of this process. The theory formally connects the likelihoods and hyperparameters across the three approaches, leverages mathematical genetics results to stabilize simplicial diffusion, and demonstrates that a single model can be trained to perform diffusion in any of the three domains at test time. Experiments report that the Wright-Fisher simplicial variant is more stable and outperforms prior simplicial models on conditional DNA generation, while multi-domain models remain competitive with single-domain baselines.
Significance. If the claimed exact equivalences hold, the unification would let practitioners interchange diffusion paradigms without retraining and borrow numerical-stability techniques from the mathematical-genetics literature. The multi-domain training result is practically attractive for applications involving DNA, proteins, or language. The work’s main contribution is the theoretical linkage rather than new algorithms, so its significance rests on the tightness of the Wright-Fisher correspondence and the reproducibility of the reported performance gains.
major comments (2)
- [Unification theory] Abstract and unification theory: the claim that standard discrete diffusion transition kernels match Wright-Fisher multinomial sampling exactly (required for identical marginal likelihoods and interchangeable hyperparameters) must be verified by direct comparison of the forward noising kernels, rate matrices, and absorbing-state handling. Any discretization mismatch would break the single-model multi-domain training justification.
- [Large-population limits] Large-population limits derivations: while the Gaussian and simplicial limits are standard diffusion approximations, the manuscript must show that the specific discretizations and stochastic processes used in existing models correspond exactly to instances of Wright-Fisher dynamics without extra approximations that would invalidate the claimed equivalence of likelihoods and hyperparameters.
minor comments (1)
- [Experiments] The abstract states that Wright-Fisher simplicial diffusion outperforms prior simplicial models on DNA generation, but the experimental section should include explicit protocol details, baseline implementations, and statistical significance tests to allow independent verification.
Simulated Author's Rebuttal
We thank the referee for the detailed and constructive review. The comments highlight important points about the rigor of the claimed equivalences, which we address by strengthening explicit comparisons and derivations in the revised manuscript. We believe these changes preserve the core contribution while improving clarity and verifiability.
read point-by-point responses
-
Referee: [Unification theory] Abstract and unification theory: the claim that standard discrete diffusion transition kernels match Wright-Fisher multinomial sampling exactly (required for identical marginal likelihoods and interchangeable hyperparameters) must be verified by direct comparison of the forward noising kernels, rate matrices, and absorbing-state handling. Any discretization mismatch would break the single-model multi-domain training justification.
Authors: We appreciate the referee's emphasis on explicit verification. The original manuscript derives the equivalence by showing that the discrete diffusion forward process is exactly the multinomial sampling step of the Wright-Fisher model under the chosen parameterization (with mutation rates governing the absorbing-state behavior). In the revision we have added a dedicated subsection (Section 3.2) that tabulates the forward noising kernels side-by-side, compares the infinitesimal rate matrices, and confirms that the absorbing-state handling is identical via the standard population-genetics mutation operator. These direct comparisons establish that the marginal likelihoods coincide exactly and that hyperparameters transfer without adjustment, thereby justifying the multi-domain training result. No discretization mismatch exists under the model definitions used. revision: yes
-
Referee: [Large-population limits] Large-population limits derivations: while the Gaussian and simplicial limits are standard diffusion approximations, the manuscript must show that the specific discretizations and stochastic processes used in existing models correspond exactly to instances of Wright-Fisher dynamics without extra approximations that would invalidate the claimed equivalence of likelihoods and hyperparameters.
Authors: We agree that the large-population limits must be shown to align precisely with the discretizations employed in prior Gaussian and simplicial diffusion models. The revised manuscript augments the derivations in Section 4 with explicit statements that the time-discretized Ornstein-Uhlenbeck process recovered in the Gaussian limit and the Dirichlet-multinomial process recovered in the simplicial limit are obtained directly from the Wright-Fisher generator without additional approximations beyond the classical large-N diffusion limit (citing the standard convergence theorems from mathematical genetics). We further verify that the noise schedules and step sizes used in the literature correspond one-to-one to the Wright-Fisher time parameterization, preserving the exact equivalence of likelihoods and hyperparameters. These additions eliminate any ambiguity about extraneous approximations. revision: yes
Circularity Check
No circularity: unification rests on external Wright-Fisher model from mathematical genetics
full rationale
The paper presents discrete, Gaussian, and simplicial diffusion as different parameterizations of the pre-existing Wright-Fisher population genetics process, with the latter two arising as large-population limits. This connection is explicitly grounded in decades of external mathematical genetics literature rather than any internal fitting, self-definition, or self-citation chain. The abstract states the theory 'formally connects the likelihoods and hyperparameters' by leveraging that literature to stabilize simplicial diffusion, and demonstrates multi-domain training without reducing any claimed equivalence to a tautology or fitted input. No load-bearing step reduces by construction to the paper's own inputs; the derivation is self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Discrete, Gaussian, and simplicial diffusion processes correspond to instances or large-population limits of the Wright-Fisher population genetics model
Reference graph
Works this paper leans on
-
[1]
S. Alamdari, N. Thakkar, R. van den Berg, A. X. Lu, N. Fusi, A. P. Amini, and K. K. Yang. Protein generation with evolutionary diffusion: sequence is all you need.bioRxiv, Sept. 2023
work page 2023
-
[2]
A. N. Amin, N. Gruver, and A. G. Wilson. Why masking diffusion works: Condition on the jump schedule for improved discrete diffusion. InFrontiers in Probabilistic Inference: Learning meets Sampling, Apr. 2025
work page 2025
-
[3]
B. D. O. Anderson. Reverse-time diffusion equation models.Stoch. Process. Their Appl., 12(3): 313–326, May 1982
work page 1982
- [4]
-
[5]
P. Avdeyev, C. Shi, Y . Tan, K. Dudnyk, and J. Zhou. Dirichlet diffusion score model for biological sequence generation.arXiv [cs.LG], May 2023
work page 2023
- [6]
-
[7]
R. F. Bass.Stochastic Processes. Cambridge University Press, Oct. 2011
work page 2011
- [8]
-
[9]
D. Calderon, R. Blecher-Gonen, X. Huang, S. Secchia, J. Kentro, R. M. Daza, B. Martin, A. Dulja, C. Schaub, C. Trapnell, E. Larschan, K. M. O’Connor-Giles, E. E. M. Furlong, and J. Shendure. The continuum of <i>drosophila</i> embryonic development at single- cell resolution.Science, 377(6606):eabn5800, 2022. doi: 10.1126/science.abn5800. URL https://www.s...
-
[10]
A. Campbell, J. Benton, V . De Bortoli, T. Rainforth, G. Deligiannidis, and A. Doucet. A continuous time framework for discrete denoising models. InAdvances in Neural Information Processing Systems, Oct. 2022
work page 2022
-
[11]
N. A. Chandra, Y . Hu, J. D. Buenrostro, S. Mostafavi, and A. Sasse. Refining sequence-to- activity models by increasing model resolution.bioRxiv, 2025. doi: 10.1101/2025.01.24.634804
- [12]
-
[13]
S. Dieleman, L. Sartran, A. Roshannai, N. Savinov, Y . Ganin, P. H. Richemond, A. Doucet, R. Strudel, C. Dyer, C. Durkan, C. Hawthorne, R. Leblond, W. Grathwohl, and J. Adler. Continuous diffusion for categorical data.arXiv.org, 2022
work page 2022
-
[14]
F. Eijkelboom, G. Bartosh, C. Andersson Naesseth, M. Welling, and J.-W. van de Meent. Variational flow matching for graph generation.Advances in Neural Information Processing Systems, 37:11735–11764, 2024
work page 2024
-
[15]
S. N. Ethier and T. G. Kurtz.Markov Processes: Characterisation and Convergence. Probability & Mathematical Statistics S. John Wiley & Sons, Nashville, TN, May 1986
work page 1986
- [16]
-
[17]
F. Gotze. On the rate of convergence in the multivariate CLT.Ann. Probab., 19(2):724–739, 1991
work page 1991
-
[18]
R. C. Griffiths. Asymptotic line-of-descent distributions.J. Math. Biol., 21(1):67–75, Dec. 1984
work page 1984
- [19]
-
[20]
X. Han, S. Kumar, and Y . Tsvetkov. SSD-LM: Semi-autoregressive simplex-based diffusion language model for text generation and modular control.arXiv [cs.CL], Oct. 2022
work page 2022
-
[21]
B. L. Hie, V . R. Shanker, D. Xu, T. U. J. Bruun, P. A. Weidenbacher, S. Tang, W. Wu, J. E. Pak, and P. S. Kim. Efficient evolution of human antibodies from general protein language models. Nat. Biotechnol., 42(2):275–283, Apr. 2023
work page 2023
-
[22]
J. Ho, A. Jain, and P. Abbeel. Denoising diffusion probabilistic models. In H. Larochelle, M. Ranzato, R. Hadsell, M. F. Balcan, and H. Lin, editors,Advances in Neural Information Processing Systems, volume 33, pages 6840–6851. Curran Associates, Inc., 2020
work page 2020
-
[23]
F. M. Hoppe. Polya-like urns and the ewens’ sampling formula.J. Math. Biol., 20(1):91–94, Aug. 1984
work page 1984
-
[24]
P. A. Jenkins and D. Spanò. Exact simulation of the Wright–Fisher diffusion.Ann. Appl. Probab., 27(3):1478–1509, June 2017
work page 2017
-
[25]
F. Johansson and Others.mpmath: a Python library for arbitrary-precision floating-point arithmetic (version 0.14), Feb. 2010
work page 2010
-
[26]
D. D. Johnson, J. Austin, R. van den Berg, and D. Tarlow. Beyond in-place corruption: Insertion and deletion in denoising probabilistic models. InICML Workshop on Invertible Neural Networks, Normalizing Flows, and Explicit Likelihood Models, 2021
work page 2021
-
[27]
M. Kimura. Solution of a process of random genetic drift with a continuous model.Proc. Natl. Acad. Sci. U. S. A., 41(3):144–150, Mar. 1955
work page 1955
-
[28]
B. Li, Z. Gao, and L. Xu. Unifying continuous and discrete text diffusion with non-simultaneous diffusion processes.arXiv [cs.CL], May 2025
work page 2025
-
[29]
Z. Li, Y . Ni, G. Xia, W. Beardall, A. Das, G.-B. Stan, and Y . Zhao. Absorb & escape: Overcoming single model limitations in generating heterogeneous genomic sequences.Advances in Neural Information Processing Systems, 37:21949–21978, 2024
work page 2024
-
[30]
Z. Lin, H. Akin, R. Rao, B. Hie, Z. Zhu, W. Lu, N. Smetanin, R. Verkuil, O. Kabeli, Y . Shmueli, A. dos Santos Costa, M. Fazel-Zarandi, T. Sercu, S. Candido, and A. Rives. Evolutionary-scale 12 prediction of atomic-level protein structure with a language model.Science, 379(6637):1123– 1130, 2023. doi: 10.1126/science.ade2574. URL https://www.science.org/d...
- [31]
-
[32]
A. Lou, C. Meng, and S. Ermon. Discrete diffusion modeling by estimating the ratios of the data distribution. In41 st International Conference on Machine Learning, Oct. 2023
work page 2023
-
[33]
S. Luo, Y . Su, X. Peng, S. Wang, J. Peng, and J. Ma. Antigen-specific antibody design and optimization with diffusion-based generative models for protein structures. InAdvances in Neural Information Processing Systems 35. Cold Spring Harbor Laboratory, July 2022
work page 2022
-
[34]
R. K. Mahabadi, H. Ivison, J. Tae, J. Henderson, I. Beltagy, M. E. Peters, and A. Cohan. TESS: Text-to-text self-conditioned simplex diffusion.arXiv [cs.CL], May 2023
work page 2023
-
[35]
J. W. Miller. Asymptotic normality, concentration, and coverage of generalized posteriors. arXiv [math.ST], July 2019
work page 2019
-
[36]
J. Ou, S. Nie, K. Xue, F. Zhu, J. Sun, Z. Li, and C. Li. Your absorbing discrete diffusion secretly models the conditional distributions of clean data.arXiv [cs.LG], June 2024
work page 2024
- [37]
-
[38]
P. H. Richemond, S. Dieleman, and A. Doucet. Categorical SDEs with simplex diffusion.arXiv [cs.LG], Oct. 2022
work page 2022
-
[39]
H. Robbins. A remark on stirling’s formula.Am. Math. Mon., 62(1):26, Jan. 1955
work page 1955
-
[40]
S. S. Sahoo, M. Arriola, Y . Schiff, A. Gokaslan, E. Marroquin, J. T. Chiu, A. Rush, and V . Kuleshov. Simple and effective masked diffusion language models.arXiv [cs.CL], June 2024
work page 2024
-
[41]
S. S. Sahoo, J. Deschenaux, A. Gokaslan, G. Wang, J. Chiu, and V . Kuleshov. The diffusion duality.arXiv [cs.LG], June 2025
work page 2025
- [42]
-
[43]
A. Shabalin, V . Meshchaninov, and D. Vetrov. Smoothie: Smoothing diffusion on token embeddings for text generation.arXiv [cs.CL], May 2025
work page 2025
-
[44]
J. Shi, K. Han, Z. Wang, A. Doucet, and M. K. Titsias. Simplified and generalized masked diffusion for discrete data.arXiv [cs.LG], June 2024
work page 2024
- [45]
-
[46]
C. Stone. Limit theorems for random walks, birth and death processes, and diffusion processes. Illinois J. Math., 7(4):638–660, Dec. 1963
work page 1963
-
[47]
B. E. Suzek, H. Huang, P. McGarvey, R. Mazumder, and C. H. Wu. UniRef: comprehensive and non-redundant UniProt reference clusters.Bioinformatics, 23(10):1282–1288, May 2007
work page 2007
-
[48]
S. Tang, Y . Zhang, A. Tong, and P. Chatterjee. Gumbel-softmax flow matching with straight- through guidance for controllable biological sequence generation.arXiv [cs.LG], Mar. 2025
work page 2025
-
[49]
S. Tavaré. Line-of-descent and genealogical processes, and their applications in population genetics models.Theor. Popul. Biol., 26(2):119–164, Oct. 1984
work page 1984
-
[50]
A. W. van der Vaart.Asymptotic Statistics. 1998. 13
work page 1998
- [51]
-
[52]
X. Wang, Z. Zheng, F. Ye, D. Xue, S. Huang, and Q. Gu. DPLM-2: A multimodal diffusion protein language model.arXiv [cs.LG], Oct. 2024
work page 2024
-
[53]
L. Winkler, L. Richter, and M. Opper. Bridging discrete and continuous state spaces: Exploring the ehrenfest process in time-continuous diffusion models.arXiv [stat.ML], May 2024
work page 2024
-
[54]
R. Wu, F. Ding, R. Wang, R. Shen, X. Zhang, S. Luo, C. Su, Z. Wu, Q. Xie, B. Berger, J. Ma, and J. Peng. High-resolutionde novostructure prediction from primary sequence.bioRxiv, page 2022.07.21.500999, July 2022
work page 2022
-
[55]
K. K. Yang, N. Fusi, and A. X. Lu. Convolutions are competitive with transformers for protein sequence pretraining.Cell Systems, 15(3):286–294, 2024
work page 2024
-
[56]
Diffusion Models are Evolutionary Algorithms
Y . Zhang, B. Hartl, H. Hazan, and M. Levin. Diffusion models are evolutionary algorithms. arXiv preprint arXiv:2410.02543, 2024
work page internal anchor Pith review Pith/arXiv arXiv 2024
-
[57]
K. Zheng, Y . Chen, H. Mao, M.-Y . Liu, J. Zhu, and Q. Zhang. Masked diffusion models are secretly time-agnostic masked models and exploit inaccurate categorical sampling.arXiv [cs.LG], Sept. 2024. 14 A Extended related work We add more related work beyond those in Sec. 2. Classical theories unifying discrete and continuous stochastic processesThere is a ...
work page 2024
-
[58]
has a similar idea, swapping the softmax for an asymmetric transformation and Gaussian diffusion with reflected Gaussian diffusion. With these simplifications however, the process is exactly (reflected) Gaussian diffusion except the input to the neural network is transformed onto a simplex; in particular, it doesn’t interact with the topology of the simpl...
work page 2025
-
[59]
Below we simply assume that1is not orthogonal to the top eigenspace ofΛ. Decompose Λ“ηVdiagp ⃗λ{ηqV T for a matrix VPR Bˆr with orthonormal columns, a vector λ of eigenvalues, and a scalar ηąmax i λi to be chosen later. For an orthonormal matrix UPR rˆr to be chosen later, define ˜V“ « Vdiagp ⃗λ{ηq1{2 UpI´diagp ⃗λ{ηqq1{2 ff so ˜V has orthonormal columns. ...
-
[60]
(Convergence of marginals)⃗ xζ t ⇝⃗ zt for eacht. 37
-
[61]
(Local uniform convergence of conditionals) Conditional distributions exist such that for each ⃗ vPRr, săt , and bounded compactly supported measurable function f, there is an ϵą0 , such that sup }⃗ w´⃗ v}ăϵ |E⃗ xζ t |⃗ xζ s “⃗ wf´E ⃗ zt|⃗ zs“⃗ wf| Ñ0
-
[62]
(Tightness) For every ra, bs Ă p0,1q , there are β, θ, Mą0 such that for all s, tP ra, bs , supζąM E}⃗ xζ s ´⃗ xζ t }β ăCps´tq θ. Then, with the topology of convergence on compact sets11, the paths converge in distribution p⃗ xζ t qtPp0,1q ⇝p⃗ ztqtPp0,1q. Proof. Pick a compact set ra, bs Ă p0,1q . We show p⃗ xζ t qtPra,bs ⇝p⃗ ztqtPra,bs. Say p⃗ xζm t qtPr...
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.