PASPT2: a size-extensive and size-consistent partial-active-space multi-state multi-reference second-order perturbation theory for strongly correlated electrons
Pith reviewed 2026-05-16 21:44 UTC · model grok-4.3
The pith
PASPT2 achieves strict size-extensivity in multi-state multi-reference perturbation theory by linearizing IN-GMS-SU-CCSD and using a reference-specific zeroth-order Hamiltonian.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
PASPT2 is obtained by linearizing IN-GMS-SU-CCSD. The disconnected terms that appear in the parent amplitude equations are eliminated completely through the choice of a special reference-specific zeroth-order Hamiltonian. The corresponding effective or intermediate Hamiltonian is then connected and closed, so that the energies produced by its diagonalization are fully connected. Consequently PASPT2 is strictly size-extensive, in contrast to IN-GMS-SU-CCSD, and is size-consistent whenever the partial active space of a supermolecule is the direct product of the partial active spaces of its physically separated, non-interacting fragments.
What carries the argument
The reference-specific zeroth-order Hamiltonian that removes disconnected terms from the linearized PASPT2 amplitude equations and renders the effective Hamiltonian connected.
If this is right
- Energies obtained by diagonalization of the effective Hamiltonian are fully connected.
- PASPT2 is strictly size-extensive, unlike the parent IN-GMS-SU-CCSD.
- PASPT2 is size-consistent for non-interacting fragments when their partial active spaces multiply directly.
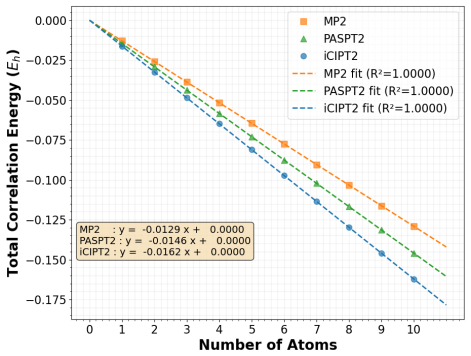
- The approach applies to prototypical strongly correlated systems and demonstrates practical efficacy.
Where Pith is reading between the lines
- The same linearization-plus-special-Hamiltonian construction could be tested at higher perturbation orders while preserving connectivity.
- Size-extensivity opens the door to direct application on extended molecular aggregates without artificial corrections for fragment separation.
- The method supplies a concrete route for embedding multi-reference perturbation theory inside a coupled-cluster parent while retaining polynomial scaling.
Load-bearing premise
Disconnected terms in the PASPT2 amplitude equations can be avoided completely by choosing a special reference-specific zeroth-order Hamiltonian.
What would settle it
Explicit calculation of the energy of two distant non-interacting fragments that shows a non-zero size-consistency error when their partial active spaces are not chosen as a direct product, or a zero error when they are.
Figures









read the original abstract
A partial-active-space (PAS) multi-state (MS) multi-reference second-order perturbation theory (MRPT2) for the electronic structure of strongly correlated systems of electrons, dubbed PASPT2, is formulated by linearizing the intermediate normalization-based general-model-space state-universal coupled-cluster theory with singles and doubles [IN-GMS-SU-CCSD; J. Chem. Phys. 119, 5320 (2003)]. At variance with the existence of disconnected terms in the IN-GMS-SU-CCSD amplitude equations, the disconnected terms in the PASPT2 amplitude equations can be avoided completely by choosing a special reference-specific zeroth-order Hamiltonian. The corresponding effective/intermediate Hamiltonian can also be made connected and closed, so as to render the energies obtained by diagonalization fully connected. As such, PASPT2 is strictly size-extensive, in sharp contrast with the parent IN-GMS-SU-CCSD. It is also size-consistent when the PAS of a supermolecule is chosen to be the direct product of those of the physically separated, non-interacting fragments. Prototypical systems are taken as showcases to reveal the efficacy of PASPT2.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript formulates PASPT2 by linearizing the intermediate-normalization general-model-space state-universal coupled-cluster singles-and-doubles (IN-GMS-SU-CCSD) equations. It asserts that a specially chosen reference-specific zeroth-order Hamiltonian eliminates all disconnected terms from the amplitude equations and renders the effective/intermediate Hamiltonian connected and closed, thereby making the method strictly size-extensive (unlike its parent) and size-consistent when the partial active space of a supermolecule is the direct product of the fragment spaces.
Significance. If the central construction holds, PASPT2 would supply a rare size-extensive and size-consistent multi-reference second-order perturbation theory for strongly correlated electrons. This addresses a long-standing limitation of multi-reference methods and could enable reliable calculations on extended systems where size-extensivity is essential. The explicit use of a reference-specific H0 to enforce connectedness is a notable technical feature, though its generality requires verification.
major comments (1)
- [Derivation of amplitude equations and effective Hamiltonian] The claim that the special reference-specific zeroth-order Hamiltonian removes all disconnected terms and yields a connected effective Hamiltonian (abstract and derivation sections) is load-bearing for both the size-extensivity and size-consistency assertions. The manuscript must explicitly demonstrate that this H0 remains additive for a supermolecule whose PAS is the direct product of the fragment PASs; otherwise non-additive components would generate disconnected contributions upon expansion of the supermolecule equations, undermining the size-consistency guarantee.
minor comments (1)
- [Numerical results section] The abstract states that prototypical systems are used as showcases; the manuscript should include a brief table or figure summarizing the size-consistency tests (e.g., energy additivity errors for separated fragments) to make the numerical support for the central claim immediately visible.
Simulated Author's Rebuttal
We thank the referee for the careful reading and constructive feedback on our manuscript. The major comment raises an important point about explicitly verifying the additivity of the reference-specific zeroth-order Hamiltonian, which we address below by strengthening the derivation.
read point-by-point responses
-
Referee: [Derivation of amplitude equations and effective Hamiltonian] The claim that the special reference-specific zeroth-order Hamiltonian removes all disconnected terms and yields a connected effective Hamiltonian (abstract and derivation sections) is load-bearing for both the size-extensivity and size-consistency assertions. The manuscript must explicitly demonstrate that this H0 remains additive for a supermolecule whose PAS is the direct product of the fragment PASs; otherwise non-additive components would generate disconnected contributions upon expansion of the supermolecule equations, undermining the size-consistency guarantee.
Authors: We agree that an explicit demonstration of additivity is required to fully substantiate the size-consistency claim. The reference-specific H0 in PASPT2 is defined as the diagonal one-electron operator whose eigenvalues are the orbital energies obtained from the reference-specific Fock operator (constructed from the density of the given reference determinant). For a supermolecule AB with PAS equal to the direct product of fragment PASs, the model-space determinants factorize as products of fragment determinants, and the underlying orbitals remain localized on each fragment. Consequently, the reference-specific orbital energies for AB are strictly additive (epsilon_i^{AB} = epsilon_i^A + epsilon_i^B for corresponding orbitals), making H0^{AB} = H0^A + H0^B exactly. This additivity ensures that the linearized amplitude equations contain no non-additive (hence potentially disconnected) contributions when the fragments are non-interacting. In the revised manuscript we have inserted a new subsection (Sec. II.C) that spells out this factorization argument, together with a short algebraic proof that the effective Hamiltonian remains connected under the same conditions. A brief numerical verification for a separated H2...H2 dimer has also been added to the results section. revision: yes
Circularity Check
Size-extensivity follows by construction from special reference-specific H0 choice that eliminates disconnected terms
specific steps
-
self definitional
[Abstract]
"At variance with the existence of disconnected terms in the IN-GMS-SU-CCSD amplitude equations, the disconnected terms in the PASPT2 amplitude equations can be avoided completely by choosing a special reference-specific zeroth-order Hamiltonian. The corresponding effective/intermediate Hamiltonian can also be made connected and closed, so as to render the energies obtained by diagonalization fully connected. As such, PASPT2 is strictly size-extensive, in sharp contrast with the parent IN-GMS-SU-CCSD. It is also size-consistent when the PAS of a supermolecule is chosen to be the direct product"
PASPT2 is defined by linearizing the parent theory plus the explicit selection of a special H0 whose sole stated purpose is to avoid disconnected terms; the size-extensivity conclusion is then asserted 'as such' from that same choice. The property is therefore equivalent to the input definition rather than independently derived.
full rationale
The paper's derivation begins with linearization of the cited IN-GMS-SU-CCSD parent method and then introduces a specially chosen reference-specific zeroth-order Hamiltonian explicitly to remove disconnected terms from the amplitude equations. The abstract directly states that this choice allows the effective Hamiltonian to be made connected, 'so as to render the energies... fully connected. As such, PASPT2 is strictly size-extensive'. The central extensivity and size-consistency claims therefore reduce directly to the definitional properties engineered into the H0 choice rather than emerging from an independent first-principles argument or external benchmark. The self-citation to the 2003 parent framework supplies the starting point but does not itself establish the new connectivity property.
Axiom & Free-Parameter Ledger
axioms (2)
- standard math Standard assumptions of many-body perturbation theory and intermediate normalization in electronic structure calculations.
- ad hoc to paper A special reference-specific zeroth-order Hamiltonian can be chosen to make all disconnected terms vanish and render the effective Hamiltonian connected.
Lean theorems connected to this paper
-
IndisputableMonolith/Cost/FunctionalEquation.leanwashburn_uniqueness_aczel echoes?
echoesECHOES: this paper passage has the same mathematical shape or conceptual pattern as the Recognition theorem, but is not a direct formal dependency.
the disconnected terms in the PASPT2 amplitude equations can be avoided completely by choosing a special reference-specific zeroth-order Hamiltonian... PASPT2 is strictly size-extensive
-
IndisputableMonolith/Foundation/ArithmeticFromLogic.leanembed_injective echoes?
echoesECHOES: this paper passage has the same mathematical shape or conceptual pattern as the Recognition theorem, but is not a direct formal dependency.
the effective/intermediate Hamiltonian can also be made connected and closed
What do these tags mean?
- matches
- The paper's claim is directly supported by a theorem in the formal canon.
- supports
- The theorem supports part of the paper's argument, but the paper may add assumptions or extra steps.
- extends
- The paper goes beyond the formal theorem; the theorem is a base layer rather than the whole result.
- uses
- The paper appears to rely on the theorem as machinery.
- contradicts
- The paper's claim conflicts with a theorem or certificate in the canon.
- unclear
- Pith found a possible connection, but the passage is too broad, indirect, or ambiguous to say the theorem truly supports the claim.
Reference graph
Works this paper leans on
-
[1]
Ruedenberg, K.; Cheung, L. M.; Elbert, S. T. MCSCF optimization through combined use of natural orbitals and the Brillouin-Levy-Berthier theorem. Int. J. Quantum Chem. 1979, 16, 1069--1101
work page 1979
-
[2]
Roos, B. O.; Taylor, P. R. A complete active space method (CASSCF) using a density matrix formulated super-CI approach. Chem. Phys. 1980, 48, 157--173
work page 1980
-
[3]
Werner, H.; Meyer, W. A quadratically convergent multiconfiguration–self‐consistent field method with simultaneous optimization of orbitals and CI coefficients. J. Chem. Phys. 1980, 73, 2342--2356
work page 1980
-
[4]
Siegbahn, P. E. M.; Almlöf, J.; Heiberg, A.; Roos, B. O. The complete active space SCF (CASSCF) method in a Newton–Raphson formulation with application to the HNO molecule. J. Chem. Phys. 1981, 74, 2384--2396
work page 1981
-
[5]
A quadratically convergent MCSCF method for the simultaneous optimization of several states
Werner, H.; Meyer, W. A quadratically convergent MCSCF method for the simultaneous optimization of several states. J. Chem. Phys. 1981, 74, 5794--5801
work page 1981
-
[6]
The effective interaction in nuclei and its perturbation expansion: An algebraic approach
Schucan, T.; Weidenm \"u ller, H. The effective interaction in nuclei and its perturbation expansion: An algebraic approach. Ann. Phys. 1972, 73, 108--135
work page 1972
-
[7]
Roos, B. O.; Andersson, K. Multiconfigurational perturbation theory with level shift—the Cr2 potential revisited. Chem. Phys. Lett. 1995, 245, 215--223
work page 1995
-
[8]
Multiconfiguration perturbation theory with imaginary level shift
Forsberg, N.; Malmqvist, P.- . Multiconfiguration perturbation theory with imaginary level shift. Chem. Phys. Lett. 1997, 274, 196--204
work page 1997
-
[9]
Hayashi, M.; Saitow, M.; Uemura, K.; Yanai, T. Quasi-degenerate extension of local N-electron valence state perturbation theory with pair-natural orbital method based on localized virtual molecular orbitals. J. Chem. Phys. 2024, 160, 194105
work page 2024
-
[10]
Li, Y.; Luo, S.; Wu, P.; Lei, Y. The application of Dyall Hamiltonian-based MRPT2 in high-lying electronically excited state calculations. Chem. Phys. Lett. 2025, 871, 142096
work page 2025
-
[11]
Bender, C. F.; Davidson, E. R. Studies in configuration interaction: The first-row diatomic hydrides. Phys. Rev. 1969, 183, 23--30
work page 1969
-
[12]
Whitten, J. L.; Hackmeyer, M. Configuration interaction studies of ground and excited states of polyatomic molecules. I. The CI formulation and studies of formaldehyde. J. Chem. Phys. 1969, 51, 5584--5596
work page 1969
-
[13]
Buenker, R. J.; Peyerimhoff, S. D. Individualized configuration selection in CI calculations with subsequent energy extrapolation. Theor. Chim. Acta 1974, 35, 33--58
work page 1974
-
[14]
Huron, B.; Malrieu, J. P.; Rancurel, P. Iterative perturbation calculations of ground and excited state energies from multiconfigurational zeroth-order wave functions. J. Chem. Phys. 1973, 58, 5745--5759
work page 1973
-
[15]
Evangelisti, S.; Daudey, J. P.; Malrieu, J. P. Convergence of an improved CIPSI algorithm. Chem. Phys. 1983, 75, 91--102
work page 1983
-
[16]
Evangelista, F. A. Adaptive multiconfigurational wave functions. J. Chem. Phys. 2014, 140, 124114
work page 2014
-
[17]
Liu, W.; Hoffmann, M. R. SDS: the `static-dynamic-static' framework for strongly correlated electrons. Theor. Chem. Acc. 2014, 133, 1481
work page 2014
-
[18]
Liu, W.; Hoffmann, M. R. iCI: Iterative CI toward full CI. J. Chem. Theory Comput. 2016, 12, 1169--1178, (E) 2016, 12, 3000
work page 2016
-
[19]
Schriber, J. B.; Evangelista, F. A. Communication: An adaptive configuration interaction approach for strongly correlated electrons with tunable accuracy. J. Chem. Phys. 2016, 144, 161106
work page 2016
-
[20]
Schriber, J. B.; Evangelista, F. A. Adaptive configuration interaction for computing challenging electronic excited states with tunable accuracy. J. Chem. Theory Comput. 2017, 13, 5354--5366
work page 2017
-
[21]
Holmes, A. A.; Tubman, N. M.; Umrigar, C. J. Heat-bath configuration interaction: an efficient selected configuration interaction algorithm inspired by heat-bath sampling. J. Chem. Theory Comput. 2016, 12, 3674--3680
work page 2016
-
[22]
Garniron, Y.; Scemama, A.; Loos, P.-F.; Caffarel, M. Hybrid stochastic-deterministic calculation of the second-order perturbative contribution of multireference perturbation theory. J. Chem. Phys. 2017, 147, 034101
work page 2017
-
[23]
Tubman, N. M.; Lee, J.; Takeshita, T. Y.; Head-Gordon, M.; Whaley, K. B. A deterministic alternative to the full configuration interaction quantum Monte Carlo method. J. Chem. Phys. 2016, 145, 044112
work page 2016
-
[24]
An efficient deterministic perturbation theory for selected configuration interaction methods
Tubman, N. M.; Levine, D. S.; Hait, D.; Head-Gordon, M.; Whaley, K. B. An efficient deterministic perturbation theory for selected configuration interaction methods. 2018, arXiv preprint arXiv:1808.02049
work page internal anchor Pith review Pith/arXiv arXiv 2018
-
[25]
Zimmerman, P. M. Incremental full configuration interaction. J. Chem. Phys. 2017, 146, 104102
work page 2017
-
[26]
Zimmerman, P. M. Strong correlation in incremental full configuration interaction. J. Chem. Phys. 2017, 146, 224104
work page 2017
-
[27]
Zhang, N.; Liu, W.; Hoffmann, M. R. Iterative Configuration Interaction with Selection. J. Chem. Theory Comput. 2020, 16, 2296--2316
work page 2020
-
[28]
Zhang, N.; Liu, W.; Hoffmann, M. R. Further Development of iCIPT2 for Strongly Correlated Electrons. J. Chem. Theory Comput. 2021, 17, 949--964
work page 2021
-
[29]
Guo, Y.; Zhang, N.; Lei, Y.; Liu, W. iCISCF: An Iterative Configuration Interaction-Based Multiconfigurational Self-Consistent Field Theory for Large Active Spaces. J. Chem. Theory Comput. 2021, 17, 7545--7561
work page 2021
-
[30]
Chilkuri, V. G.; Neese, F. Comparison of many-particle representations for selected-CI I: A tree based approach. J. Comput. Chem. 2021, 42, 982--1005
work page 2021
-
[31]
Chilkuri, V. G.; Neese, F. Comparison of many-particle representations for selected configuration interaction: II. Numerical benchmark calculations. J. Chem. Theory Comput. 2021, 17, 2868--2885
work page 2021
-
[32]
Eriksen, J. J.; Anderson, T. A.; Deustua, J. E.; Ghanem, K.; Hait, D.; Hoffmann, M. R.; Lee, S.; Levine, D. S.; Magoulas, I.; Shen, J.; Tubman, N. M.; Whaley, K. B.; Xu, E.; Yao, Y.; Zhang, N.; Alavi, A.; Chan, G. K.-L.; Head-Gordon, M.; Liu, W.; Piecuch, P.; Sharma, S.; Ten-no, S. L.; Umrigar, C. J.; Gauss, J. The Ground State Electronic Energy of Benzen...
work page 2020
-
[33]
Epstein, P. S. The stark effect from the point of view of Schroedinger's quantum theory. Phys. Rev. 1926, 28, 695
work page 1926
-
[34]
Nesbet, R. K. Configuration interaction in orbital theories. Proc. Roy. Soc. of London. Ser. A 1955, 230, 312--321
work page 1955
-
[35]
Mahapatra, U. S.; Datta, B.; Mukherjee, D. Development of a size-consistent state-specific multireference perturbation theory with relaxed model-space coefficients. Chem. Phys. Lett. 1999, 299, 42--50
work page 1999
-
[36]
Sinha Mahapatra, U.; Datta, B.; Mukherjee, D. Molecular applications of a size-consistent state-specific multireference perturbation theory with relaxed model-space coefficients. J. Phys. Chem. A 1999, 103, 1822--1830
work page 1999
-
[37]
A spin-adapted size-extensive state-specific multi-reference perturbation theory
Mao, S.; Cheng, L.; Liu, W.; Mukherjee, D. A spin-adapted size-extensive state-specific multi-reference perturbation theory. I. Formal developments. J. Chem. Phys. 2012, 136, 024105
work page 2012
-
[38]
Mao, S.; Cheng, L.; Liu, W.; Mukherjee, D. A spin-adapted size-extensive state-specific multi-reference perturbation theory with various partitioning schemes. II. Molecular applications. J. Chem. Phys. 2012, 136, 024106
work page 2012
-
[39]
Sen, A.; Sen, S.; Samanta, P. K.; Mukherjee, D. Unitary group adapted state specific multireference perturbation theory: Formulation and pilot applications. J. Comput. Chem. 2015, 36, 670--688
work page 2015
-
[40]
Li, C.; Evangelista, F. A. Multireference driven similarity renormalization group: A second-order perturbative analysis. J. Chem. Theory Comput. 2015, 11, 2097--2108
work page 2015
-
[41]
Li, C.; Evangelista, F. A. Driven similarity renormalization group for excited states: A state-averaged perturbation theory. J. Chem. Phys. 2018, 148, 124106
work page 2018
-
[42]
A Jeziorski-Monkhorst fully uncontracted multi-reference perturbative treatment
Giner, E.; Angeli, C.; Garniron, Y.; Scemama, A.; Malrieu, J.-P. A Jeziorski-Monkhorst fully uncontracted multi-reference perturbative treatment. I. Principles, second-order versions, and tests on ground state potential energy curves. J. Chem. Phys. 2017, 146, 224108
work page 2017
-
[43]
Introduction of n-electron valence states for multireference perturbation theory
Angeli, C.; Cimiraglia, R.; Evangelisti, S.; Leininger, T.; Malrieu, J.-P. Introduction of n-electron valence states for multireference perturbation theory. J. Chem. Phys. 2001, 114, 10252--10264
work page 2001
-
[44]
Angeli, C.; Cimiraglia, R.; Malrieu, J.-P. n-electron valence state perturbation theory: A spinless formulation and an efficient implementation of the strongly contracted and of the partially contracted variants. J. Chem. Phys. 2002, 117, 9138--9153
work page 2002
-
[45]
A new size extensive multireference perturbation theory
Chen, F.; Fan, Z. A new size extensive multireference perturbation theory. J. Comput. Chem. 2014, 35, 121--129
work page 2014
-
[46]
Multireference Rayleigh--Schr \"o dinger perturbation theory and its application
Yi, J.; Chen, F. Multireference Rayleigh--Schr \"o dinger perturbation theory and its application. J. Chem. Phys. 2019, 150, 124108
work page 2019
-
[47]
Rosta, E.; Surj \'a n, P. R. Two-body zeroth order Hamiltonians in multireference perturbation theory: The APSG reference state. J. Chem. Phys. 2002, 116, 878--890
work page 2002
-
[48]
F \"o ldv \'a ri, D.; T \'o th, Z.; Surj \'a n, P. R.; Szabados, \'A . Geminal perturbation theory based on the unrestricted Hartree--Fock wavefunction. J. Chem. Phys. 2019, 150, 034103
work page 2019
-
[49]
Block correlated second order perturbation theory with a generalized valence bond reference function
Xu, E.; Li, S. Block correlated second order perturbation theory with a generalized valence bond reference function. J. Chem. Phys. 2013, 139, 174111
work page 2013
-
[50]
Diagrammatic many-body perturbation theory for general model spaces
Hose, G.; Kaldor, U. Diagrammatic many-body perturbation theory for general model spaces. J. Phys. B: At. Mol. Phys. 1979, 12, 3827--3855
work page 1979
-
[51]
A general-model-space diagrammatic perturbation theory
Hose, G.; Kaldor, U. A general-model-space diagrammatic perturbation theory. Phys. Scr. 1980, 21, 357--361
work page 1980
-
[52]
Quasidegenerate perturbation theory
Hose, G.; Kaldor, U. Quasidegenerate perturbation theory. J. Phys. Chem. 1982, 86, 2133--2140
work page 1982
-
[53]
Li, X.; Paldus, J. General-model-space state-universal coupled-cluster theory: Connectivity conditions and explicit equations. J. Chem. Phys. 2003, 119, 5320--5333
work page 2003
-
[54]
Jeziorski, B.; Monkhorst, H. J. Coupled-cluster method for multideterminantal reference states. Phys. Rev. A 1981, 24, 1668--1681
work page 1981
-
[55]
Sur la th \'e orie des perturbations des \'e tats li \'e s
Bloch, C. Sur la th \'e orie des perturbations des \'e tats li \'e s. Nuc. Phys. 1958, 6, 329--347
work page 1958
-
[56]
Atomic many-body theory; Springer Science & Business Media, 2012; Vol
Lindgren, I.; Morrison, J. Atomic many-body theory; Springer Science & Business Media, 2012; Vol. 3
work page 2012
-
[57]
Andersson, K.; Malmqvist, P.-A.; Roos, B. O. Second-order perturbation theory with a complete active space self-consistent field reference function. J. Chem. Phys. 1992, 96, 1218--1226
work page 1992
-
[58]
Chaudhuri, R.; Sinha, D.; Mukherjee, D. On the extensivity of the roots of effective Hamiltonians in many-body formalisms employing incomplete model spaces. Chem. Phys. Lett. 1989, 163, 165--170
work page 1989
-
[59]
Bartlett, R. J.; Purvis, G. D. Many-body perturbation theory, coupled-pair many-electron theory, and the importance of quadruple excitations for the correlation problem. Int. J. Quantum Chem. 1978, 14, 561--581
work page 1978
-
[60]
Paldus, J.; Li, X.; Petraco, N. D. General-model-space state--universal coupled-cluster method: Diagrammatic approach. J. Math. Chem. 2004, 35, 215--251
work page 2004
-
[61]
Reflections on size-extensivity, size-consistency and generalized extensivity in many-body theory
Nooijen, M.; Shamasundar, K.; Mukherjee, D. Reflections on size-extensivity, size-consistency and generalized extensivity in many-body theory. Mol. Phys. 2005, 103, 2277--2298
work page 2005
-
[62]
M ller, C.; Plesset, M. S. Note on an approximation treatment for many-electron systems. Phys. Rev. 1934, 46, 618--622
work page 1934
-
[63]
Pople, J. A.; Binkley, J. S.; Seeger, R. Theoretical models incorporating electron correlation. Int. J. Quantum Chem. Symp. 1976, 10, 1--19
work page 1976
-
[64]
Wolinski, K.; Sellers, H. L.; Pulay, P. Consistent generalization of the M ller-Plesset partitioning to open-shell and multiconfigurational SCF reference states in many-body perturbation theory. Chem. Phys. Lett. 1987, 140, 225--231
work page 1987
-
[65]
MetaWave: A Platform for Unified Implementation of Nonrelativistic and Relativistic Wave Functions
Zhang, N.; Wang, Q.; Liu, W. MetaWave: A Platform for Unified Implementation of Nonrelativistic and Relativistic Wave Functions. J. Phys. Chem. A 2025, 129, 5170--5188
work page 2025
-
[66]
Saad, Y.; Schultz, M. H. GMRES: A generalized minimal residual algorithm for solving nonsymmetric linear systems. SIAM J. Sci. Stat. Comput. 1986, 7, 856--869
work page 1986
-
[67]
Huang, C.; Liu, W.; Xiao, Y.; Hoffmann, M. R. iVI: An iterative vector interaction method for large eigenvalue problems. J. Comput. Chem. 2017, 38, 2481--2499, (E) 2018, 39, 338
work page 2017
-
[68]
Dunning Jr, T. H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007--1023
work page 1989
-
[69]
Liu, W.; Hong, G.; Dai, D.; Li, L.; Dolg, M. The Beijing four-component density functional program package (BDF) and its application to EuO, EuS, YbO and YbS. Theor. Chem. Acc. 1997, 96, 75--83
work page 1997
-
[70]
Performance of the general-model-space state-universal coupled-cluster method
Li, X.; Paldus, J. Performance of the general-model-space state-universal coupled-cluster method. J. Chem. Phys. 2004, 120, 5890--5902
work page 2004
-
[71]
Lei, Y.; Liu, W.; Hoffmann, M. R. Further development of SDSPT2 for strongly correlated electrons. Mol. Phys. 2017, 115, 2696--2707 mcitethebibliography PASPT2org.tex0000664000000000000000000037034215122713001011762 0ustar rootroot [journal=jctcce,manuscript=article] achemso amssymb,amsmath,amsfonts,multicol,multirow,longtable,array,mathpazo lscape [versi...
work page 2017
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.