Constrained nuclear-electronic orbital second-order Moller-Plesset perturbation theory
Pith reviewed 2026-05-16 02:13 UTC · model grok-4.3
The pith
CNEO-MP2 captures nuclear vibrational effects on molecular properties in one calculation by adding electronic-nuclear correlation.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Derived from a multicomponent generalization of the Hylleraas functional, the CNEO-MP2 method includes electronic-nuclear and nuclear correlation to calculate vibrationally averaged molecular properties within the constrained nuclear-electronic orbital framework, as demonstrated by benchmarks on diatomic and small polyatomic molecules and ions.
What carries the argument
Multicomponent generalization of the Hylleraas functional, used to generate the second-order perturbation expansion inside the CNEO framework.
If this is right
- CNEO-MP2 produces internuclear distances and bond angles that already incorporate vibrational averaging.
- It generates potential energy surfaces and vibrational frequencies that reflect nuclear quantum effects directly.
- Nuclear quantum effects such as zero-point energy and isotopic shifts are obtained without additional post-processing force-constant calculations.
- The method applies to both diatomic and small polyatomic molecules and ions in a single computation.
Where Pith is reading between the lines
- The single-step workflow could lower the cost of including nuclear quantum effects when screening larger sets of molecules.
- Routine use in quantum chemistry codes might make vibrational averaging a default option rather than an optional add-on.
- The approach might be extended to compute averaged properties along reaction paths where zero-point corrections matter.
Load-bearing premise
The multicomponent generalization of the Hylleraas functional supplies a valid starting point for a second-order perturbation expansion inside the constrained nuclear-electronic orbital framework.
What would settle it
Benchmark calculations on the test set of molecules and ions showing that CNEO-MP2 internuclear distances or vibrational frequencies deviate significantly from experimental values or from established methods that include nuclear quantum effects would falsify the central claim.
Figures
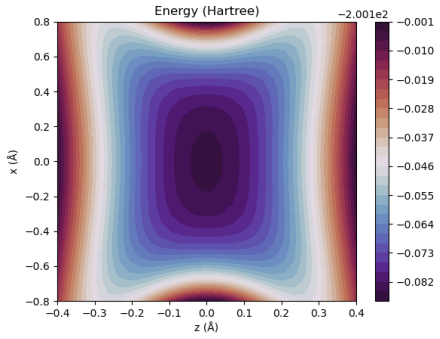
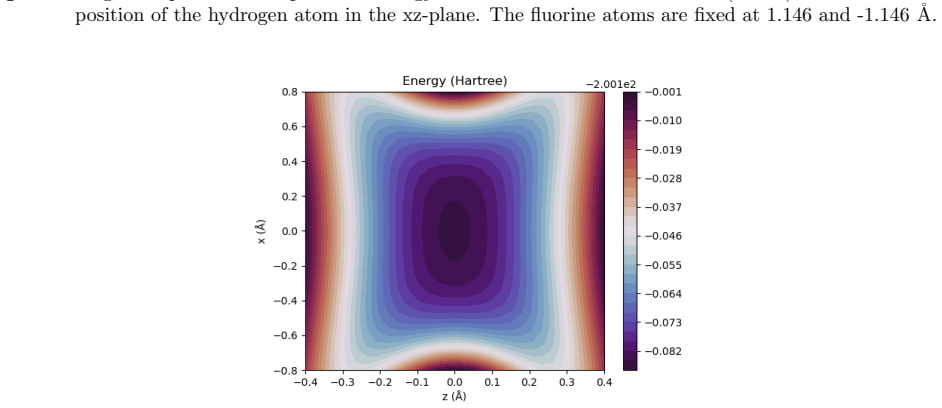
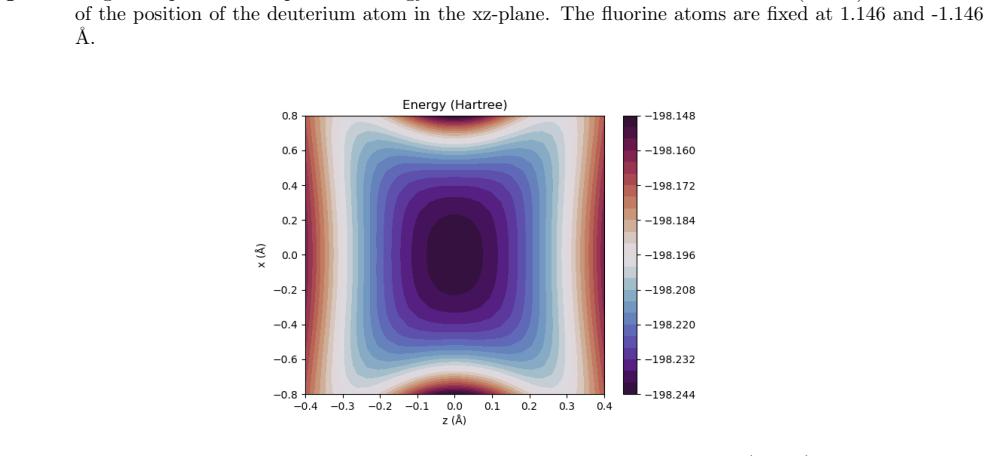
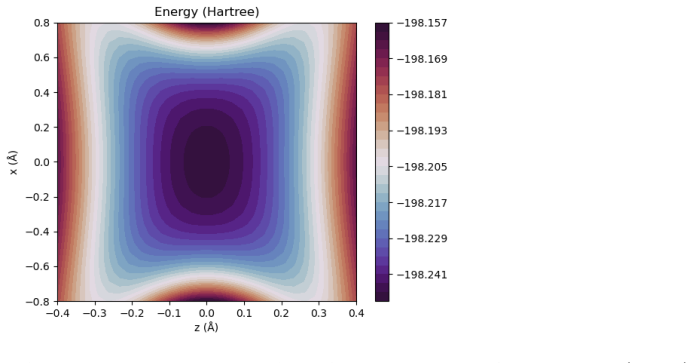
read the original abstract
A multicomponent second-order M{\o}ller-Plesset perturbation theory (MP2) method is derived and implemented within the constrained nuclear-electronic orbital (CNEO) framework from a multicomponent generalization of the Hylleraas functional. The CNEO-MP2 method includes electronic-nuclear and nuclear correlation in the calculation of vibrationally averaged molecular properties. Nuclear quantum effects like vibrational averaging, isotopic effects, and zero-point energy can be captured in a single calculation or geometry optimization with CNEO-MP2, eliminating the need to perform costly subsequent calculations to determine higher order force constants as required with many existing methods used to determine vibrational effects upon molecular properties. The CNEO-MP2 method is benchmarked on a test set of diatomic and small polyatomic molecules and ions. Herein, we present internuclear distances, bond angles, potential energy surfaces, and vibrational frequencies calculated with the CNEO-MP2 method to demonstrate that it correctly captures the effects of nuclear vibrational motion upon molecular properties.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript derives and implements a multicomponent second-order Møller-Plesset perturbation theory (CNEO-MP2) within the constrained nuclear-electronic orbital (CNEO) framework, starting from a multicomponent generalization of the Hylleraas functional. The method incorporates electronic-nuclear and nuclear correlation to compute vibrationally averaged molecular properties such as internuclear distances, bond angles, potential energy surfaces, and vibrational frequencies. Benchmarks on diatomic and small polyatomic molecules and ions are presented to demonstrate capture of nuclear quantum effects including vibrational averaging, isotopic shifts, and zero-point energy in a single calculation or geometry optimization, without requiring separate higher-order force constant evaluations.
Significance. If the central derivation is sound, the CNEO-MP2 approach offers a meaningful advance by embedding nuclear quantum effects directly into a perturbative electronic-structure framework, potentially reducing the computational overhead associated with post hoc vibrational averaging or anharmonic corrections in conventional methods. This could prove useful for systems where nuclear motion significantly influences properties, such as hydrogen-bonded complexes or isotopically sensitive reactions.
major comments (2)
- [§2.3, Eq. (12)] §2.3, Eq. (12): The multicomponent Hylleraas functional is generalized to CNEO, but the text does not explicitly introduce Lagrange multipliers for the nuclear-electronic orbital constraints or demonstrate that the first-order stationarity conditions remain satisfied after the MP2 amplitudes are solved. Without this step, it is unclear whether the second-order energy correction remains strictly within the constrained manifold or admits unphysical contributions that do not correspond to vibrational averaging.
- [§4.2, Table 2] §4.2, Table 2: The reported mean absolute deviations for vibrational frequencies (approximately 15 cm⁻¹ across the test set) are presented without direct comparison to CCSD(T) or experimental values with propagated uncertainties; this weakens the claim that CNEO-MP2 “correctly captures” the effects, as the magnitude of improvement over CNEO-HF or other baselines is not quantified.
minor comments (2)
- [§2.1] The definition of the nuclear-electronic orbital basis functions in §2.1 is introduced without an accompanying table of symbols, making it difficult to track the distinction between electronic and nuclear orbital indices throughout the amplitude equations.
- [Figure 3] Figure 3 caption states that the potential energy surface is “vibrationally averaged,” but the figure itself shows only the raw CNEO-MP2 curve; a second panel or inset comparing the averaged versus unaveraged surface would improve clarity.
Simulated Author's Rebuttal
We thank the referee for the positive assessment of the CNEO-MP2 method and its potential utility for incorporating nuclear quantum effects. We address the two major comments below with clarifications and planned revisions.
read point-by-point responses
-
Referee: [§2.3, Eq. (12)] §2.3, Eq. (12): The multicomponent Hylleraas functional is generalized to CNEO, but the text does not explicitly introduce Lagrange multipliers for the nuclear-electronic orbital constraints or demonstrate that the first-order stationarity conditions remain satisfied after the MP2 amplitudes are solved. Without this step, it is unclear whether the second-order energy correction remains strictly within the constrained manifold or admits unphysical contributions that do not correspond to vibrational averaging.
Authors: The CNEO framework enforces the nuclear-electronic orbital constraints at the mean-field (CNEO-HF) level via Lagrange multipliers that define the constrained orbitals. The multicomponent Hylleraas functional is then constructed and minimized within this fixed constrained orbital basis, so the MP2 amplitudes are solved in the space orthogonal to the constraints. This construction ensures the second-order correction remains strictly on the constrained manifold by design. We acknowledge that the manuscript does not explicitly restate the stationarity conditions after the amplitudes are obtained; we will add a short paragraph and an auxiliary equation in §2.3 of the revised manuscript to demonstrate that the first-order stationarity conditions continue to hold. revision: partial
-
Referee: [§4.2, Table 2] §4.2, Table 2: The reported mean absolute deviations for vibrational frequencies (approximately 15 cm⁻¹ across the test set) are presented without direct comparison to CCSD(T) or experimental values with propagated uncertainties; this weakens the claim that CNEO-MP2 “correctly captures” the effects, as the magnitude of improvement over CNEO-HF or other baselines is not quantified.
Authors: We agree that direct comparisons and quantification of improvement would strengthen the presentation. In the revised manuscript we will augment Table 2 (and the accompanying text) with CCSD(T) reference values for the subset of molecules where they are available, experimental vibrational frequencies, and the corresponding mean absolute deviations for CNEO-HF. This will allow explicit quantification of the improvement provided by the MP2 correlation terms. revision: yes
Circularity Check
CNEO-MP2 derivation from multicomponent Hylleraas generalization is self-contained with no reduction to inputs
full rationale
The paper derives the CNEO-MP2 method explicitly from a multicomponent generalization of the Hylleraas functional inside the CNEO framework, then benchmarks it on diatomic and polyatomic test sets for properties such as internuclear distances and vibrational frequencies. No equations or steps in the abstract or described derivation reduce a prediction to a fitted parameter, self-definition, or self-citation chain; the Hylleraas starting point is treated as an external functional whose stationarity conditions are assumed to hold under CNEO constraints, and the MP2 amplitudes are obtained by standard perturbation theory. The central claim therefore retains independent content from the functional generalization and the subsequent implementation, yielding a normal non-circular finding.
Axiom & Free-Parameter Ledger
Reference graph
Works this paper leans on
-
[1]
D. M. Bishop. Molecular motion in electric fields.Rev. Mod. Phys.1990,62(2), 343–374
work page 1990
-
[2]
R. P. A. Bettens. Bound State Potential Energy Surface Construction: Ab Initio Zero-point Energies and Vibrationally Averaged Rotational Constants.J. Am. Chem. Soc.2003,125(2), 584–587
work page 2003
-
[3]
C. Puzzarini and J. F. Stanton. Connections between the accuracy of rotational constants and equi- librium molecular structures.Phys. Chem. Chem. Phys.2023,25(3), 1421–1429. 30
work page 2023
-
[4]
P. Solomon and J. M. Schulman. The Effects of Vibrational Averaging on the Calculated Nuclear Spin–Spin Coupling Constants of Ammonia.J. Am. Chem. Soc.1977,99(24), 7776–7779
work page 1977
- [5]
-
[6]
S. Grigoleit and M. B¨ uhl. Thermal Effects and Vibrational Corrections to Transition Metal NMR Chemical Shifts.Chem. Eur. J.2004,10(21), 5541–5552
work page 2004
-
[7]
T. Tuttle. Averaging Semiempirical NMR Chemical Shifts: Dynamic Effects on the Subpicosecond Time Scale.J. Phys. Chem. A2009,113(43), 11723–11733
-
[8]
M. Draˇ c´ ınsk´ y and P. Hodgkinson. A molecular dynamics study of the effects of fast molecular motions on solid-state NMR parameters.Cryst. Eng. Comm.2013,15(43), 8705–8712
work page 2013
- [9]
- [10]
- [11]
-
[12]
X. Chen, Z. Rinkevicius, Z. Cao, K. Ruud, and H. ˚Agren. Zero-point vibrational corrections to isotropic hyperfine coupling constants in polyatomic molecules.Phys. Chem. Chem. Phys.2011,13 (2), 696–707
work page 2011
-
[13]
B. C. Mort and J. Autschbach. Magnitude of Zero-Point Vibrational Corrections to Optical Rotation in Rigid Organic Molecules: A Time-Dependent Density Functional Study.J. Phys. Chem. A2005, 109(38), 8617–8623
-
[14]
J. M. Bowman. The Self-Consistent-Field Approach to Polyatomic Vibrations.Acc. Chem. Res.1986, 19(7), 202–208
work page 1986
-
[15]
O. Christiansen. Vibrational structure theory: new vibrational wave function methods for calculation 31 of anharmonic vibrational energies and vibrational contributions to molecular properties.Phys. Chem. Chem. Phys.2007,9(23), 2942–2953
work page 2007
- [16]
-
[17]
K. R. Glaesemann and L. E. Fried. A path integral approach to molecular thermochemistry.J. Chem. Phys.2003,118(4), 1596–1603
work page 2003
-
[18]
T. L´ opez-Ciudad, R. Ram´ ırez, J. Schulte, M. C. B¨ ohm. Anharmonic effects on the structural and vibrational properties of the ethyl radical: A path integral Monte Carlo study.J. Chem. Phys.2003, 119(8), 4328–4338
work page 2003
-
[19]
H. H. Nielsen, The Vibration-Rotation Energies of Molecules.Rev. Mod. Phys.1951,23(2), 90–136
work page 1951
-
[20]
I. M. Mills. Vibrational perturbation theory.J. Mol. Spec.1961,5(1-6), 334–340
work page 1961
-
[21]
J. Neugebauer and B. A. Hess. Fundamental vibrational frequencies of small polyatomic molecules from density-functional calculations and vibrational perturbation theory.J. Chem. Phys2003,118 (16), 7215–7225
-
[22]
J. Bloino and V. Barone. A second-order perturbation theory route to vibrational averages and transition properties of molecules: General formulation and application to infrared and vibrational circular dichroism spectroscopies.J. Chem. Phys.2012,136(12), 124108-1–124108-15
work page 2012
-
[23]
P. R. Franke, J. F. Stanton, and G. E. Douberly. How to VPT2: Accurate and Intuitive Simula- tions of CH Stretching Infrared Specta Using VPT2+K with Large Effective Hamiltonian Resonance Treatments.J. Phys. Chem. A2021,125(6), 1301–1324
-
[24]
R. C. Fortenberry. Picking up Good Vibrations through Quartic Force Fields and Vibrational Per- turbation Theory.J. Phys. Chem. Lett2024,15(25), 6528–6537
- [25]
-
[26]
M. Tachikawa, K. Mori, H. Nakai, K. Iguchi. An extension of ab initio molecular orbital theory to nuclear motion.Chem. Phys. Lett.1998,290(4-6), 437–442. 32
work page 1998
-
[27]
S. Webb, T. Iordanov, and S. Hammes-Schiffer. Multiconfigurational nuclear-electronic orbital ap- proach: Incorporation of nuclear quantum effects in electronic structure calculations.J. Chem. Phys. 2002,117(9) 4106–4118
work page 2002
- [28]
-
[29]
R. U. Khan and R. Tonner-Zech. Geometrical Isotope Effects on Chemical Bonding in Hydrogen Bonded Systems: Combining Nuclear-Electronic Orbital DFT and Energy Decomposition Analysis. J. Comput. Chem.2025,46(24), 1–12
work page 2025
-
[30]
L. E. Smith, V. Briega-Martos, Y. Yang, and S. Hammes-Schiffer. Isotope Effects for Water at Pt(111) Computed with Nuclear-Electronic Orbital Theory.J. Chem. Phys. Lett.2025,16(51), 13054–13061
work page 2025
-
[31]
I. L. Thomas. Protonic Structure of Molecules. I. Ammonia Molecules.Phys. Rev.1969,185(1), 90–94
work page 1969
-
[32]
I. L. Thomas. Protonic Structure of Molecules. II. Methodology, Center-of-Mass Transformation, and the Structure of Methane, Ammonia, and Water.Phys. Rev. A1970,2(4), 1200–1208
-
[33]
H. Nakai. Simultaneous Determination of Nuclear and Electronic Wave Functions Without Born- Oppenheimer Approximation: Ab Initio NO+MO/HF Theory.Int. J. Quant. Chem.2002,86(6), 511–517
work page 2002
-
[34]
H. Nakai. Nuclear Orbital Plus Molecular Orbital Theory: Simultaneous Determination of Nuclear and Electronic Wave Functions Without Born–Oppenheimer Approximation.Int. J. Quant. Chem. 2007,107(14), 2849–2869
work page 2007
-
[35]
F. Pavoˇ sevi´ c, T. Culpitt, and S. Hammes-Schiffer. Multicomponent Quantum Chemistry: Integrating Electronic and Nuclear Quantum Effects via the Nuclear–Electronic Orbital Method.Chem. Rev.2020, 120(9), 4222–4253
work page 2020
-
[36]
M. V. Pak, A. Chakraborty, and S. Hammes-Schiffer. Density Functional Theory Treatment of Elec- tron Correlation in the Nuclear-Electronic Orbital Approach.J. Phys. Chem. A2007,111(20), 4522–4526
-
[37]
A. Chakraborty, M. V. Pak, and S. Hammes-Schiffer. Properties of the exact universal functional in multicomponent density functional theory.J. Chem. Phys.2009,131(12), 124115-1–124115-8. 33
work page 2009
-
[38]
A. Chakraborty, M. V. Pak, and S. Hammes-Schiffer. Development of Electron-Proton Density Func- tionals for Multicomponent Density Functional Theory.Phys. Rev. Lett.2008,101(15), 153001- 1–153001-4; Erratum:Phys. Rev. Lett.2011,106(16), 169902-1
work page 2008
-
[39]
Y. Yang, K. R. Brorsen, T. Culpitt, M. V. Pak, and S. Hammes-Schiffer. Development of a prac- tical multicomponent density functional for electron-proton correlation to produce accurate proton densities.J. Chem. Phys.2017,147(11), 114113-1–114113-5
work page 2017
-
[40]
K. R. Brorsen, Y. Yang, and S. Hammes-Schiffer. Multicomponent Density Functional Theory: Im- pact of Nuclear Quantum Effects on Proton Affinities and Geometries.J. Chem. Phys. Lett.2017,8 (15), 3488–3493
work page 2017
-
[41]
K. R. Brorsen, P. E. Schneider, and S. Hammes-Schiffer. Alternative forms and transferability of electron-proton correlation functionals in nuclear-electronic orbital density functional theory.J. Chem. Phys.2018,149(4), 044110-1 –044110-7
work page 2018
-
[42]
Y. Yang, T. Culpitt, and S. Hammes-Schiffer. Multicomponent Time-Dependent Density Functional Theory: Proton and Electron Excitation Energies.J. Phys. Chem. Lett.2018,9(7), 1765–1770
work page 2018
-
[43]
T. Culpitt, Y. Yang, F. Pavoˇ sevi´ c, Z. Tao, and S. Hammes-Schiffer. Enhancing the applicability of multicomponent time-dependent density functional theory.J. Chem. Phys.2019,150(20), 201101- 1–201101-6
work page 2019
-
[44]
Z. Tao, Y. Yang, and S. Hammes-Schiffer. Multicomponent density functional theory: Including the density gradient in the electron-proton correlation functional for hydrogen and deuterium.J. Chem. Phys.2019,151(12), 124102-1–124102-7
work page 2019
-
[45]
Q.Yu and S. Hammes-Schiffer. Nuclear-Electronic Orbital Multistate Density Functional Theory.J. Phys. Chem. Lett.2020,11(23), 10106–10113
work page 2020
-
[46]
L. Hasecke and R. A. Mata. Multicomponent Double-Hybrid Density Functional Theory.J. Chem. Theory Comput.2025,21(22), 11509–11520
work page 2025
-
[47]
M. Gimferrer, L. Hasecke, M. B¨ odecker, and R. A. Mata. Accurate vibrational hydrogen-bond shift predictions with multicomponent DFT.Chem. Sci.2025,16(24), 11002–11011. 34
work page 2025
- [48]
-
[49]
H. Nakai and K. Sodeyama. Many-body effects in nonadiabatic molecular theory for simultaneous determination of nuclear and electronic wave functions:Ab initioNOMO/MBPT and CC methods. J. Chem. Phys.2003,118(3), 1119–1127
work page 2003
-
[50]
T. Iordanov and S. Hammes-Schiffer. Vibrational analysis for the nuclear–electronic orbital method. J. Chem. Phys.2003,118(21), 9489–9496
work page 2003
-
[51]
C. Swalina, M. V. Pak, and S. Hammes-Schiffer. Alternative formulation of many-body perturbation theory for electron–proton correlation.Chem. Phys. Lett.2005,404(4–6), 394–399
work page 2005
-
[52]
J. H. Skone, M. V. Pak, and S. Hammes-Schiffer. Nuclear-electronic orbital nonorthogonal configu- ration interaction approach.J. Chem. Phys.2005,123(13), 134108-1–134108-8
work page 2005
-
[53]
B. Auer and S. Hammes-Schiffer. Localized Hartree product treatment of multiple protons in the nuclear-electronic orbital framework.J. Chem. Phys.2010,132(8), 084110-1–084110-8
work page 2010
-
[54]
F. Pavoˇ sevi´ c, T. Culpitt, and S. Hammes-Schiffer. Multicomponent Coupled Cluster Singles and Doubles Theory within the Nuclear-Electronic Orbital Framework.J. Chem. Theory Comp.2019,15 (1), 338–347
work page 2019
-
[55]
F. Pavoˇ sevi´ c and S. Hammes-Schiffer. Multicomponent coupled cluster singles and doubles and Brueckner doubles methods: Proton densities and energies.J. Chem. Phys.2019,151(7), 074104- 1–074104-7
work page 2019
-
[56]
F. Pavoˇ sevi´ c, B. J. G. Rousseau, and S. Hammes-Schiffer. Multicomponent Orbital-Optimized Per- turbation Theory Methods: Approaching Coupled Cluster Accuracy at Lower Cost.J. Phys. Chem. Lett.2020,11(4), 1578–1583
work page 2020
-
[57]
O. J. Fajen and K. R. Brorsen. Separation of electron-electron and electron-proton correlation in mul- ticomponent orbital-optimized perturbation theory.J. Chem. Phys2020,152(19), 194107-1–194107-8
-
[58]
F. Pavoˇ sevi´ c, Z. Tao, and S. Hammes-Schiffer. Multicomponent Coupled Cluster Singles and Doubles with Density Fitting: Protonated Water Tetramers with Quantized Protons.J. Phys. Chem. Lett. 2021,12(6), 1631–1637. 35
work page 2021
-
[59]
N. Alaal and K. R. Brorsen. Multicomponent heat-bath configuration interaction with the perturba- tive correction for the calculation of protonic excited states.J. Chem. Phys.2021,155(23), 234107- 1–234107-11
work page 2021
-
[60]
O. J. Fajen, and K. R. Brorsen. Multicomponent MP4 and the inclusion of triple excitations in multicomponent many-body methods.J. Chem. Phys.2021,155(23), 234108-1–234108-9
work page 2021
-
[61]
D. Fowler and K. R. Brorsen. (T) Correction for Multicomponent Coupled-Cluster Theory for a Single Quantum Proton.J. Chem. Theory Comput.2022,18(12), 7298–7305. Erratum:J. Chem. Theory Comput.2025,21(9), 5005–5007
work page 2022
-
[62]
L. Hasecke and R. A. Mata. Nuclear Quantum Effects Made Accessible: Local Density Fitting in Multicomponent Methods.J. Chem. Theory Comput.2023,19(22), 8223–8233
work page 2023
-
[63]
L. Hasecke and R. A. Mata. Local Electronic Correlation in Multicomponent Møller-Plesset Pertur- bation Theory.J. Chem. Theory Comput.2023,20(22), 9928–9938
work page 2023
-
[64]
R. J. Goudy, F. Pavoˇ sevi´ c, and S. Hammes-Schiffer. Triple excitations in nuclear–electronic orbital coupled cluster theory for multiple quantum protons.J. Chem. Phys.2025,163(22), 224119-1–224119- 8
work page 2025
-
[65]
J. H. Fetherholf, F. Pavoˇ sevi´ c, and S. Hammes-Schiffer. Nuclear-electronic orbital second-order cou- pled cluster for excited states.arXiv Preprint2025, 1–10
-
[66]
R. M. Nieminen, E. Boro´ nski, and L. J. Lantto. Two-component density functional theory: Applica- tion to positron states.Phys. Rev. B1985,32(2), 1377–1379
- [67]
-
[68]
E. Boro´ nski and R. M. Nieminen. Electron-positron density-functional theory.Phys. Rev. B1986, 34(6), 3820–3831
-
[69]
D. V. Makhov and L. J. Lewis. Two-component density functional theory calculations of positron lifetimes for small vacancy clusters in silicon.Phys. Rev. B2005,71(20), 205215-1–205215-6. 36
-
[70]
M. Goli and S. Shahbazian. Two-component density functional theory for muonic molecules: Inclusion of the electron–positive muon correlation functional.J. Chem. Phys.2022,156(4), 044104-1–044104- 16
work page 2022
-
[71]
N. S. Riyahi, M. Goli, and S. Shahbazian. Quantifying errors of electron-proton/muon correlation functionals through the Kohn-Sham inversion of a two-component model system.Phys. Rev. B2023, 108(24), 245155-1–245155-20
-
[72]
T. Udagawa, T. Tsuneda, and M. Tachikawa. Electron-nucleus correlation functional for multicom- ponent density functional thoery.Phys. Rev. A2014,89(5), 052519-1–052519-6
-
[73]
Z. Pan, L. Peng, Z. Pan, Y. Yuan, H. Zhang, and B. Ye. F-µbond length andµSR depolarization spectrum calculation for fluoride using two-component density functional theory.Chinese Phys. B 2023,32(8), 087602-1–087602-4
work page 2023
- [74]
-
[75]
M. Goli and S. Shahbazian. Developing effective electronic-only coupled-cluster and Møller-Plesset perturbation theories for the muonic molecules.Phys. Chem. Chem. Phys.2018,20(24), 16749–16760
work page 2018
- [76]
-
[77]
B. H. Ellis, S. Aggarway, and A. Chakraborty. Development of the Multicomponent Coupled-Cluster Theory for Investigation of Multiexcitonic Interactions.J. Chem. Theory Comput.2016,12(1), 188–200
work page 2016
- [78]
-
[79]
K. Miyamoto, M. Hoshino, and H. Nakai. Elimination of Translational and Rotational Motions in Nu- clear Orbital Plus Molecular Orbital Theory: Contribution of the First-Order Rovibration Coupling. J. Chem. Theory Comput.2006,2(6), 1544–1550. 37
work page 2006
-
[80]
A. D. Bochevearov, E. F. Valeev, and C. D. Sherrill. The electron and nuclear orbitals model: current challenges and future prospectsMol. Phys.2006,102(1), 111–123
work page 2006
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.