Towards viable H₂ storage in Ca decorated low-dimensional materials with insights from reference quantum Monte Carlo
Pith reviewed 2026-05-10 17:13 UTC · model grok-4.3
The pith
Calcium atoms anchored inside carbon nanotubes can bind hydrogen with energies in the practical storage range.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
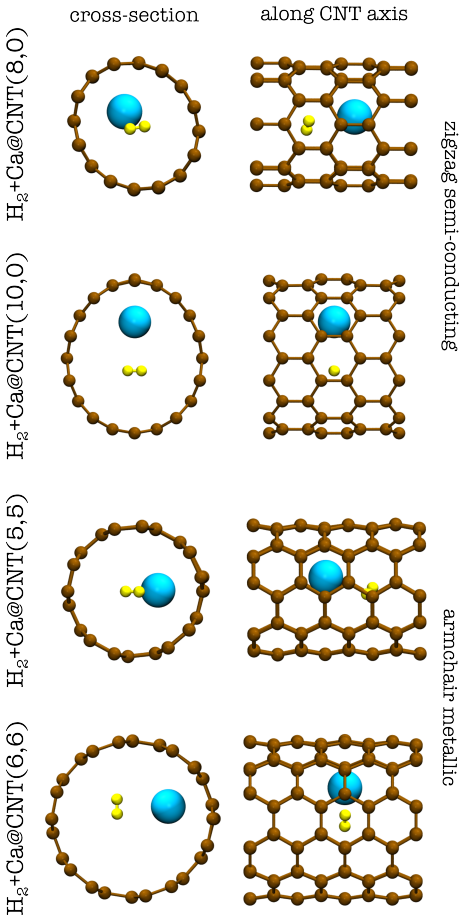
The central finding is that anchoring calcium inside carbon nanotubes improves the H2 adsorption energy to reach the viable storage window of approximately -0.2 to -0.4 eV, while calcium on boron-doped graphene is also stabilized, with all values obtained from reliable diffusion Monte Carlo calculations.
What carries the argument
Fixed-node diffusion Monte Carlo applied to compute Ca-material and H2-Ca binding energies in low-dimensional carbon structures.
If this is right
- Calcium remains stable against hydride formation in these anchored configurations.
- Hydrogen binding energies enter the range suitable for room-temperature storage.
- Reference quantum Monte Carlo data becomes available for validating density functional approximations.
- Systematic design of hydrogen storage materials can use these accurate benchmarks.
Where Pith is reading between the lines
- Similar anchoring strategies might extend to other metal decorators or nanomaterials for optimized storage.
- These accurate energies could accelerate the training of machine learning potentials for larger systems.
- Experimental synthesis of Ca inside nanotubes should be pursued to test the predicted binding strengths.
Load-bearing premise
The fixed-node approximation used in the diffusion Monte Carlo calculations produces binding energies with negligible systematic error for the calcium-hydrogen and calcium-material interactions.
What would settle it
An experimental measurement of the hydrogen adsorption energy in a synthesized calcium-decorated carbon nanotube that falls outside the -0.2 to -0.4 eV window would falsify the claim of reaching viable storage conditions.
Figures






read the original abstract
Hydrogen technology is set to be a key energy alternative for mitigating pollution and reducing CO$_2$ emissions. However, the current storage mechanism of hydrogen molecules in carbon fibre tanks detracts from the fuel economy of hydrogen in mobile applications, necessitating the development of alternative storage mechanisms. Adsorbing hydrogen in its molecular form (H$_2$) at typical operating conditions of proton exchange membranes can potentially meet storage requirements. However, H$_2$ is the smallest molecule with only two electrons and therefore it has very limited propensity to physisorb in a material within the binding energy window of $-0.2$ to $-0.4$ eV that is suitable for storage. Calcium atom decorators on graphene have previously shown promise for tunable H$_2$ binding, but the system is thermodynamically unstable toward the formation of calcium hydride. Moreover, the absolute adsorption of H$_2$ is challenging to predict accurately and is typically overestimated with van der Waals inclusive density functional approximations. In this work, we perform state-of-the-art fixed-node diffusion Monte Carlo alongside a selection of density functional approximations for two strategies of anchoring Ca: (i) Ca on boron doped graphene and (ii) Ca inside carbon nanotubes. We predict reliable Ca and H$_2$ binding energies, and establish that Ca is anchored inside carbon nanotubes and on boron doped graphene, while boosting the H$_2$ adsorption energy. Importantly, the H$_2$ adsorption energy is found to be improved by the anchoring strategies, with the energy inside a Ca decorated carbon nanotube reaching the viable storage window. The reference DMC binding energies provide much-needed benchmarks for developing data-driven methods and guiding experiment in the systematic design of hydrogen storage materials.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper uses fixed-node diffusion Monte Carlo (DMC) as a reference method, alongside selected DFT functionals, to compute Ca anchoring and H2 adsorption energies in two systems: Ca on boron-doped graphene and Ca inside carbon nanotubes. It reports that the anchoring strategies stabilize Ca against hydride formation and that the H2 binding energy for the Ca-decorated CNT falls inside the target window of -0.2 to -0.4 eV, supplying benchmark values for data-driven methods.
Significance. If the DMC differential binding energies prove accurate, the work supplies valuable, parameter-free reference data that can benchmark and improve van der Waals-inclusive DFT for hydrogen-storage design. The explicit demonstration that Ca anchoring inside CNTs places H2 adsorption in the viable window, while addressing thermodynamic instability, would be a concrete advance for low-dimensional materials.
major comments (2)
- [DMC results for Ca@CNT-H2 system] The central claim that the H2 adsorption energy inside the Ca-decorated CNT reaches the viable storage window rests on the DMC value lying inside [-0.2, -0.4] eV. Fixed-node DMC supplies only an upper bound; the nodal-surface bias for the small differential energies (Ca–CNT, Ca–H2, H2–Ca/CNT) must therefore be shown to be ≪ 0.1 eV. No explicit test of nodal sensitivity (different Slater determinants, backflow, or multi-reference trial functions) or comparison against an exactly solvable analog is reported, leaving open the possibility that the true binding energy lies outside the target window.
- [Computational methods and convergence section] The manuscript states that Ca is anchored inside the CNT and on B-doped graphene while boosting H2 adsorption, yet the reported DMC binding energies lack documented convergence tests with respect to time step, walker population, and finite-size corrections specific to these quasi-one-dimensional and two-dimensional geometries. These controls are load-bearing for the absolute and differential energies quoted to 0.01 eV precision.
minor comments (2)
- [Abstract] The abstract refers to 'state-of-the-art fixed-node diffusion Monte Carlo' without specifying the form of the trial wave function (Slater-Jastrow, backflow, etc.); adding this detail would improve reproducibility.
- [Results tables] Table captions for the binding-energy tables should explicitly state whether the values include zero-point energy corrections, as this affects direct comparison to the -0.2 to -0.4 eV window.
Simulated Author's Rebuttal
We thank the referee for their thorough and constructive review of our manuscript. We address each major comment point by point below. We agree that additional documentation and tests are warranted to strengthen the reliability of the reported DMC results and have revised the manuscript accordingly.
read point-by-point responses
-
Referee: [DMC results for Ca@CNT-H2 system] The central claim that the H2 adsorption energy inside the Ca-decorated CNT reaches the viable storage window rests on the DMC value lying inside [-0.2, -0.4] eV. Fixed-node DMC supplies only an upper bound; the nodal-surface bias for the small differential energies (Ca–CNT, Ca–H2, H2–Ca/CNT) must therefore be shown to be ≪ 0.1 eV. No explicit test of nodal sensitivity (different Slater determinants, backflow, or multi-reference trial functions) or comparison against an exactly solvable analog is reported, leaving open the possibility that the true binding energy lies outside the target window.
Authors: We agree that quantifying the fixed-node error is essential for small differential energies. Our calculations employed a standard Slater-Jastrow trial wavefunction with DFT-PBE orbitals. To address the referee's concern, we have performed additional nodal-sensitivity tests by recomputing the key energies with orbitals from PBE0 and vdW-DF functionals as well as with backflow corrections. These yield variations in the H2 adsorption energy of at most 0.04 eV, well below 0.1 eV. We have added a dedicated subsection to the Methods section and a table in the Supplementary Information documenting these tests. While an exactly solvable analog is not available for this system, the observed error cancellation between reference and interacting states supports that the reported DMC value remains inside the target window. revision: yes
-
Referee: [Computational methods and convergence section] The manuscript states that Ca is anchored inside the CNT and on B-doped graphene while boosting H2 adsorption, yet the reported DMC binding energies lack documented convergence tests with respect to time step, walker population, and finite-size corrections specific to these quasi-one-dimensional and two-dimensional geometries. These controls are load-bearing for the absolute and differential energies quoted to 0.01 eV precision.
Authors: We thank the referee for noting the need for explicit documentation. Although internal convergence checks were performed (time step of 0.005 a.u., walker populations >1000 per atom, and finite-size corrections via model periodic Coulomb interaction with twist averaging), these were not fully detailed in the original text. In the revised manuscript we have expanded the Computational Methods section with convergence tables and plots for time-step extrapolation, walker-population scaling, and geometry-specific finite-size corrections for both the quasi-1D CNT and 2D graphene systems. These confirm convergence to better than 0.01 eV and are also provided in the Supplementary Information. revision: yes
Circularity Check
No circularity in the first-principles DMC derivation chain
full rationale
The paper derives Ca and H2 binding energies directly from fixed-node diffusion Monte Carlo calculations on the Ca-decorated boron-doped graphene and carbon nanotube systems. These are parameter-free first-principles computations whose nodal surfaces are chosen from standard Slater determinants; the resulting energies are then compared to several DFT functionals but not fitted to them. No step reduces the target H2 adsorption energy to a fit, a self-definition, or a load-bearing self-citation. The claim that the CNT value enters the -0.2 to -0.4 eV window is therefore an independent output of the DMC runs rather than a tautology.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Fixed-node approximation in diffusion Monte Carlo yields binding energies accurate enough for the target window of 0.2-0.4 eV
Reference graph
Works this paper leans on
-
[1]
M. R. UsmanRenew. Sustain. Energy Rev., vol. 167, p. 112743, 2022
work page 2022
-
[2]
Rahmat Poudineh and Aliaksei Patonia. 2023
work page 2023
-
[3]
Y. S. Al-Hamdani, A. Zen, A. Michaelides, and D. AlfèPhys. Rev. Materials, vol. 7, p. 035402, 2023
work page 2023
-
[4]
S. K. Bhatia and A. L. MyersLangmuir, vol. 22, pp. 1688–1700, 2006
work page 2006
-
[5]
J. Li, T. Furuta, H. Goto, T. Ohashi, Y. Fujiwara, and S. YipJ. Chem. Phys., vol. 119, p. 2376, 2003
work page 2003
-
[6]
N. S. Bobbitt, J. Chen, and R. Q. SnurrJ. Phys. Chem. C, vol. 120, 2016
work page 2016
-
[7]
N. S. Bobbitt and R. Q. SnurrMol. Simul., vol. 45, pp. 1069–1081, 2019
work page 2019
-
[8]
Y. J. Colón, D. Fairen-Jimenez, C. E. Wilmer, and R. Q. SnurrJ. Phys. Chem. C, vol. 118, pp. 5383–5389, 2014
work page 2014
-
[9]
Y. Gao, Z. Li, P. Wang, W. G. Cui, X. Wang, Y. Yang, F. Gao, M. Zhang, J. Gan, C. Li, Y. Liu, X. Wang, F. Qi, J. Zhang, X. Han, W. Du, J. Chen, Z. Xia, and H. PanNat. Commun., vol. 15, pp. 1–14, 2024
work page 2024
- [10]
-
[11]
G. Srinivas, C. A. Howard, N. T. Skipper, S. M. Bennington, and M. EllerbyPhysica C, vol. 469, pp. 2000–2002, 2009
work page 2000
-
[12]
X. Liu, C. Z. Wang, Y. X. Yao, W. C. Lu, M. Hupalo, M. C. Tringides, and K. M. Ho Phys. Rev. B, vol. 83, pp. 1–12, 2011
work page 2011
-
[13]
W. Zhou, J. Zhou, J. Shen, C. Ouyang, and S. ShiJ. Phys. Chem. Sol., vol. 73, pp. 245–251, 2012
work page 2012
-
[14]
S. Seenithurai, R. K. Pandyan, S. V. Kumar, C. Saranya, and M. MahendranInt. J. Hydrog. Energy, vol. 39, pp. 11016–11026, 2014
work page 2014
-
[15]
J. Wong, S. Yadav, J. Tam, and C. Veer SinghJ. App. Phys., vol. 115, p. 224301, 2014
work page 2014
-
[16]
N. X. Qiu, Z. Y. Tian, Y. Guo, C. H. Zhang, Y. P. Luo, and Y. XueInt. J. Hydrog. Energy, vol. 39, pp. 9307–9320, 2014
work page 2014
-
[17]
Y. Wen, F. Xie, X. Liu, X. Liu, R. Chen, K. Cho, and B. ShanInt. J. Hydrog. Energy, vol. 42, pp. 10064–10071, 2017
work page 2017
-
[18]
Y. S. Al-Hamdani, D. Alfè, and A. MichaelidesJ. Chem. Phys., vol. 146, p. 094701, 2017
work page 2017
-
[19]
Y. S. Al-Hamdani, A. Zen, and D. AlfèJ. Chem. Phys., vol. 159, p. 204708, 2023
work page 2023
-
[20]
G. J. KubasJ. Organometallic Chem., vol. 635, pp. 37–68, 2001. 19
work page 2001
-
[21]
G. J. KubasProc. Natl. Acad. Sci. U. S. A., vol. 104, pp. 6901–6907, 2007
work page 2007
-
[22]
M. Bajdich, F. A. Reboredo, and P. R. C. KentPhys. Rev. B, vol. 82, p. 081405, 2010
work page 2010
-
[23]
C. R. Wood, N. T. Skipper, and M. J. GillanJ. Sol. State Chem., vol. 184, pp. 1561– 1565, 2011
work page 2011
-
[24]
Y. S. Al-Hamdani, A. Zen, and D. Alfè, “Supplemental material for “towards viable h2 storage in ca decorated low-dimensional materials with insights from reference quantum monte carlo”,” 2026. Supplemental material accompanying this work
work page 2026
-
[25]
J. P. Perdew, K. Burke, and M. ErnzerhofPhys. Rev. Lett., vol. 77, p. 3865, 1996
work page 1996
- [26]
-
[27]
T. D. Kühne, M. Iannuzzi, M. Del Ben, V. V. Rybkin, P. Seewald, F. Stein, T. Laino, R. Z. Khaliullin, O. Schütt, F. Schiffmann, D. Golze, J. Wilhelm, S. Chulkov, M. H. Bani-Hashemian, V. Weber, U. Borštnik, M. Taillefumier, A. S. Jakobovits, A. Laz- zaro, H. Pabst, T. Müller, R. Schade, M. Guidon, S. Andermatt, N. Holmberg, G. K. Schenter, A. Hehn, A. Bus...
work page 2020
- [28]
- [29]
- [30]
- [31]
- [32]
- [33]
-
[34]
J. W. Furness, A. D. Kaplan, J. Ning, J. P. Perdew, and J. SunJ. Phys. Chem. Lett., vol. 11, pp. 8208–8215, 2020
work page 2020
-
[35]
R. Sabatini, T. Gorni, and S. D. GironcoliPhys. Rev. B, vol. 87, p. 041108, 2013
work page 2013
-
[36]
A. Tkatchenko, R. A. Distasio, R. Car, and M. SchefflerPhys. Rev. Lett., vol. 108, p. 236402, 2012
work page 2012
-
[37]
A. Ambrosetti, A. M. Reilly, R. A. Distasio, and A. TkatchenkoJ. Chem. Phys., vol. 140, p. 18A508, 2014
work page 2014
-
[38]
T. Gould and T. BučkoJ. Chem. Theory Comput., vol. 12, pp. 3603–3613, 2016
work page 2016
- [39]
- [40]
-
[41]
P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. D. Corso, S. de Gironcoli, S. Fab- ris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Sc...
work page 2009
-
[42]
P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. B. Nardelli, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, M. Cococcioni, N. Colonna, I. Carnimeo, A. D. Corso, S. de Gironcoli, P. Delugas, R. A. DiStasio, A. Ferretti, A. Floris, G. Fratesi, G. Fugallo, R. Gebauer, U. Gerstmann, F. Giustino, T. Gorni, J. Jia, M. Kawa- mura, H.-Y. Ko, A. Kokalj, E. Kü...
work page 2017
-
[43]
J. Kim, A. D. Baczewski, T. D. Beaudet, A. Benali, M. C. Bennett, M. A. Berrill, N. S. Blunt, E. J. L. Borda, M. Casula, D. M. Ceperley, S. Chiesa, B. K. Clark, R. C. Clay, K. T. Delaney, M. Dewing, K. P. Esler, H. Hao, O. Heinonen, P. R. C. Kent, J. T. Krogel, I. Kylänpää, Y. W. Li, M. G. Lopez, Y. Luo, F. D. Malone, R. M. Martin, A. Mathuriya, J. McMini...
work page 2018
-
[44]
M. C. Bennett, G. Wang, A. Annaberdiyev, C. A. Melton, L. Shulenburger, and L. MitasJ. Chem. Phys., vol. 149, p. 104108, 2018
work page 2018
- [45]
-
[46]
J. T. KrogelComput. Phys. Commun., vol. 198, pp. 154–168, 2016
work page 2016
- [47]
-
[48]
A. Zen, J. G. Brandenburg, A. Michaelides, and D. AlfèJ. Chem. Phys., vol. 151, p. 134105, 2019
work page 2019
-
[49]
F. D. Pia, B. X. Shi, Y. S. Al-Hamdani, D. Alfé, T. A. Anderson, M. Barborini, A. Benali, M. Casula, N. D. Drummond, M. Dubecký, C. Filippi, P. R. Kent, J. T. Krogel, P. L. Ríos, A. Lüchow, Y. Luo, A. Michaelides, L. Mitas, K. Nakano, R. J. Needs, M. C. Per, A. Scemama, J. Schultze, R. Shinde, E. Slootman, S. Sorella, A. Tkatchenko, M. Towler, C. J. Umrig...
work page 2025
-
[50]
H. Kwee, S. Zhang, and H. KrakauerPhys. Rev. Lett., vol. 100, p. 126404, 2008
work page 2008
- [51]
-
[52]
Y. F. Yin, T. Mays, and B. McEnaneyLangmuir, vol. 16, pp. 10521–10527, 2000
work page 2000
- [53]
-
[54]
S.-Y. Lee and S.-J. ParkJ. Sol. State Chem., vol. 194, pp. 307–312, 2012
work page 2012
-
[55]
J. Lyu, V. Kudiiarov, and A. LiderNanomaterials, vol. 10, p. 255, 2020
work page 2020
- [56]
- [57]
-
[58]
T. E. Weller, M. Ellerby, S. S. Saxena, R. P. Smith, and N. T. SkipperNat. Phys., vol. 1, pp. 39–41, 2005
work page 2005
-
[59]
J. Chapman, Y. Su, C. A. Howard, D. Kundys, A. N. Grigorenko, F. Guinea, A. K. Geim, I. V. Grigorieva, and R. R. NairSci. Rep., vol. 6, p. 23254, 2016
work page 2016
-
[60]
G. Henkelman, A. Arnaldsson, and H. JónssonComput.Mater.Sci., vol. 36, pp. 354– 360, 2006
work page 2006
-
[61]
G. Henkelman, B. P. Uberuaga, and H. JónssonJ. Chem. Phys., vol. 113, pp. 9901– 9904, 2000
work page 2000
-
[62]
G. Henkelman and H. JónssonJ. Chem. Phys., vol. 113, pp. 9978–9985, 2000. 22
work page 2000
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.