Tailoring the Optoelectronic, Photocatalytic, Thermoelectric and Thermodynamic Properties of Halides Li2InBiX6 (X = Cl, Br, I) for Energy Conversion: A DFT Study
Pith reviewed 2026-05-10 17:13 UTC · model grok-4.3
The pith
Li2InBiX6 double perovskite halides are stable direct-bandgap semiconductors with tunable properties for optoelectronic, thermoelectric, and photocatalytic energy conversion.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
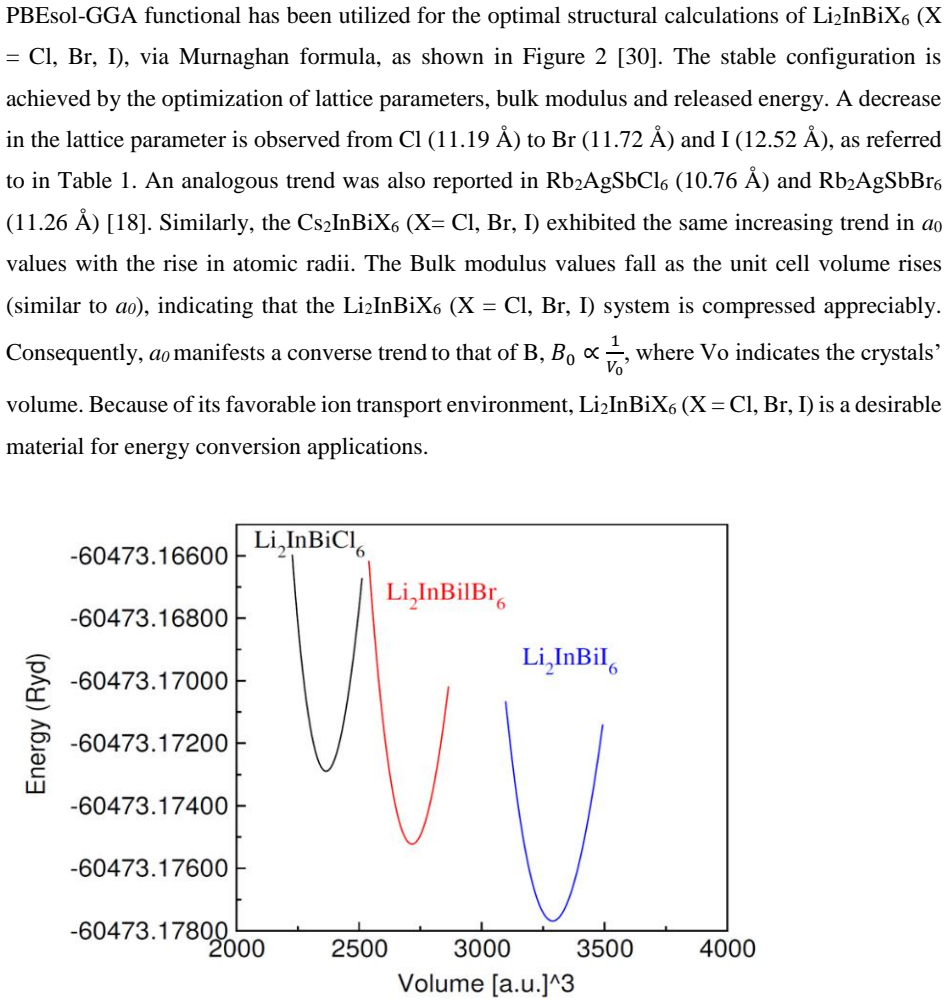
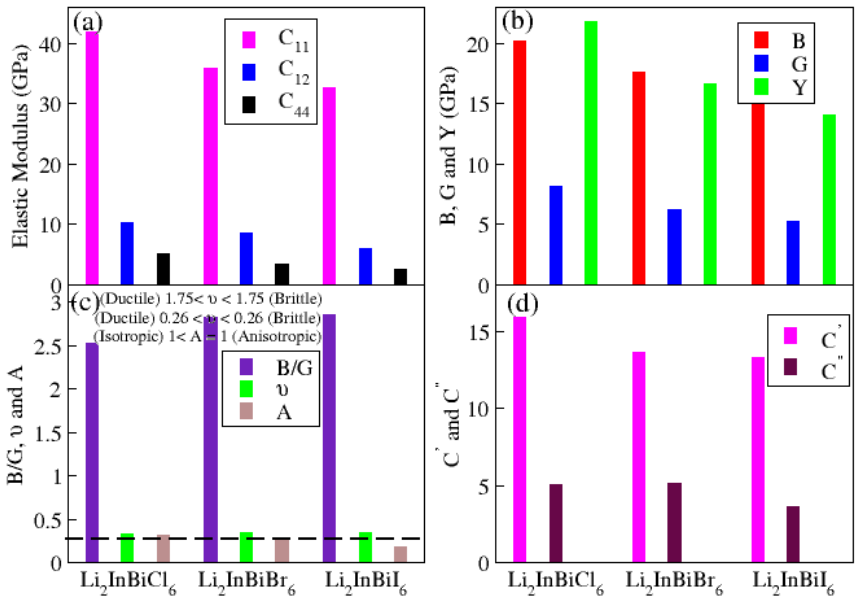
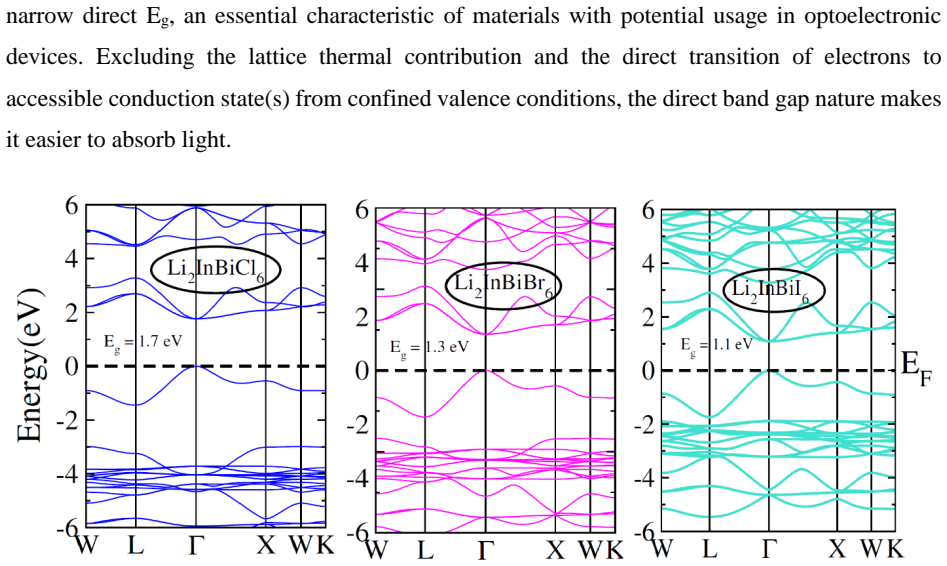
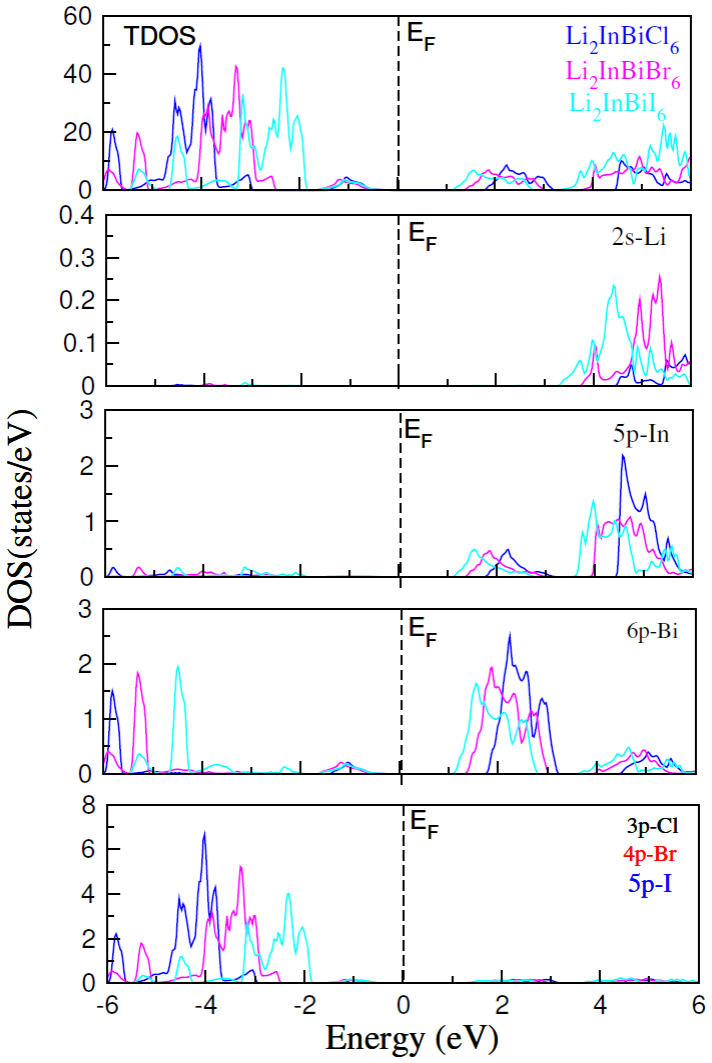
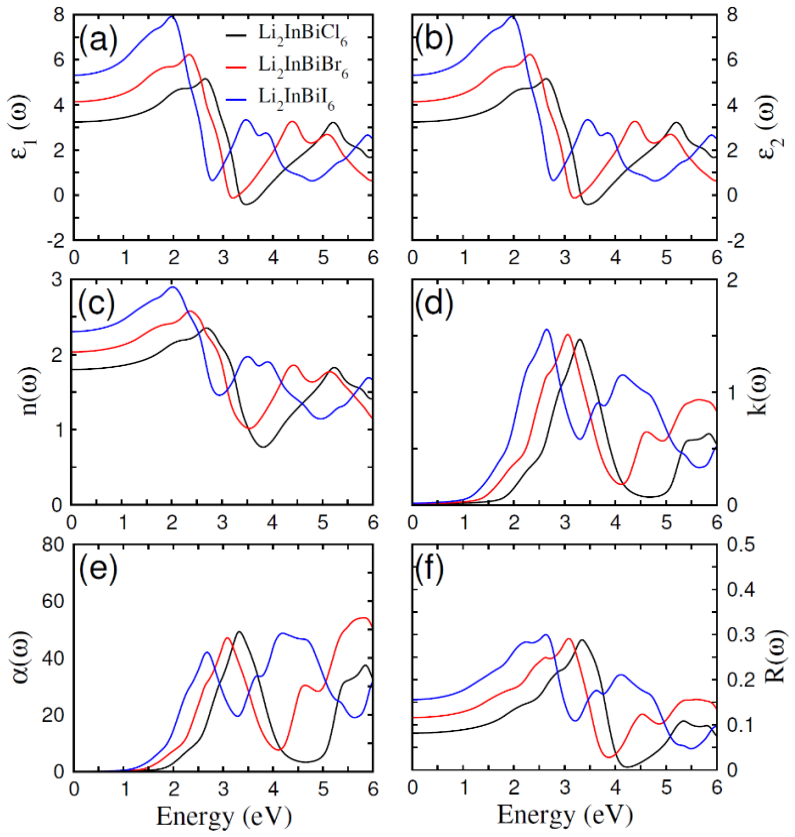
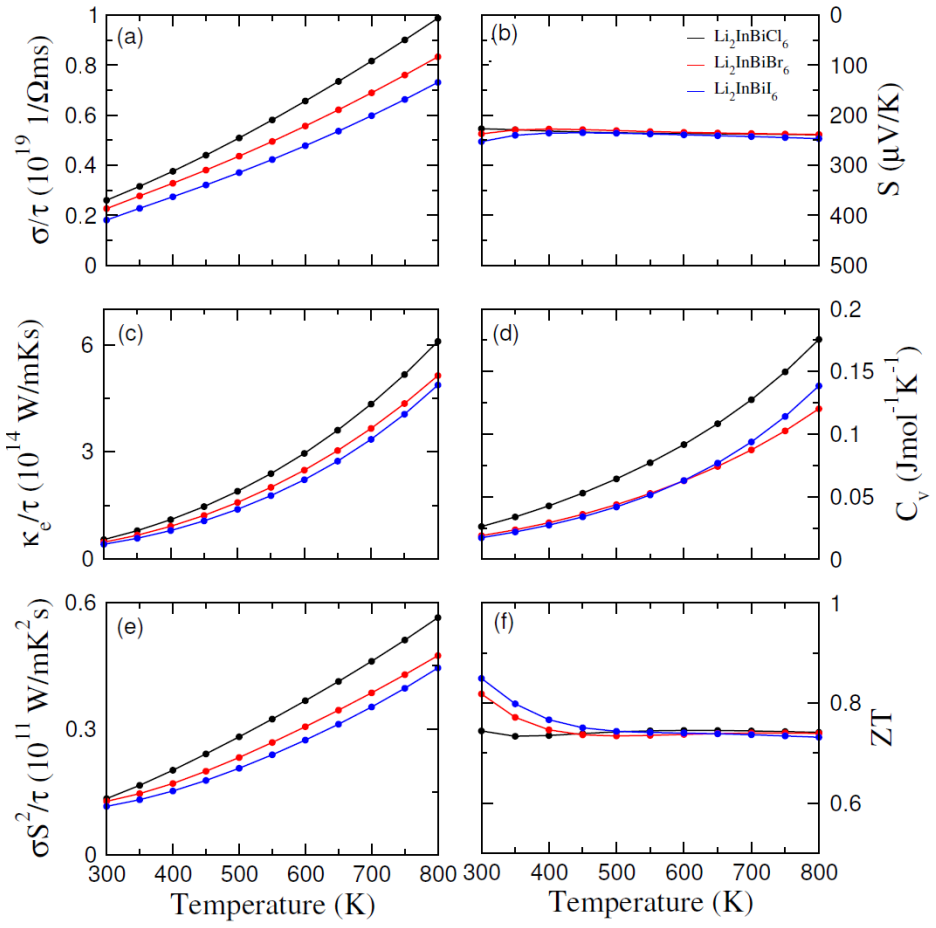
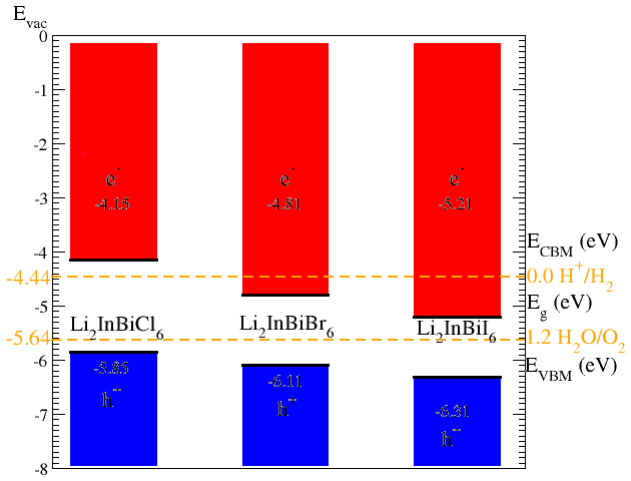
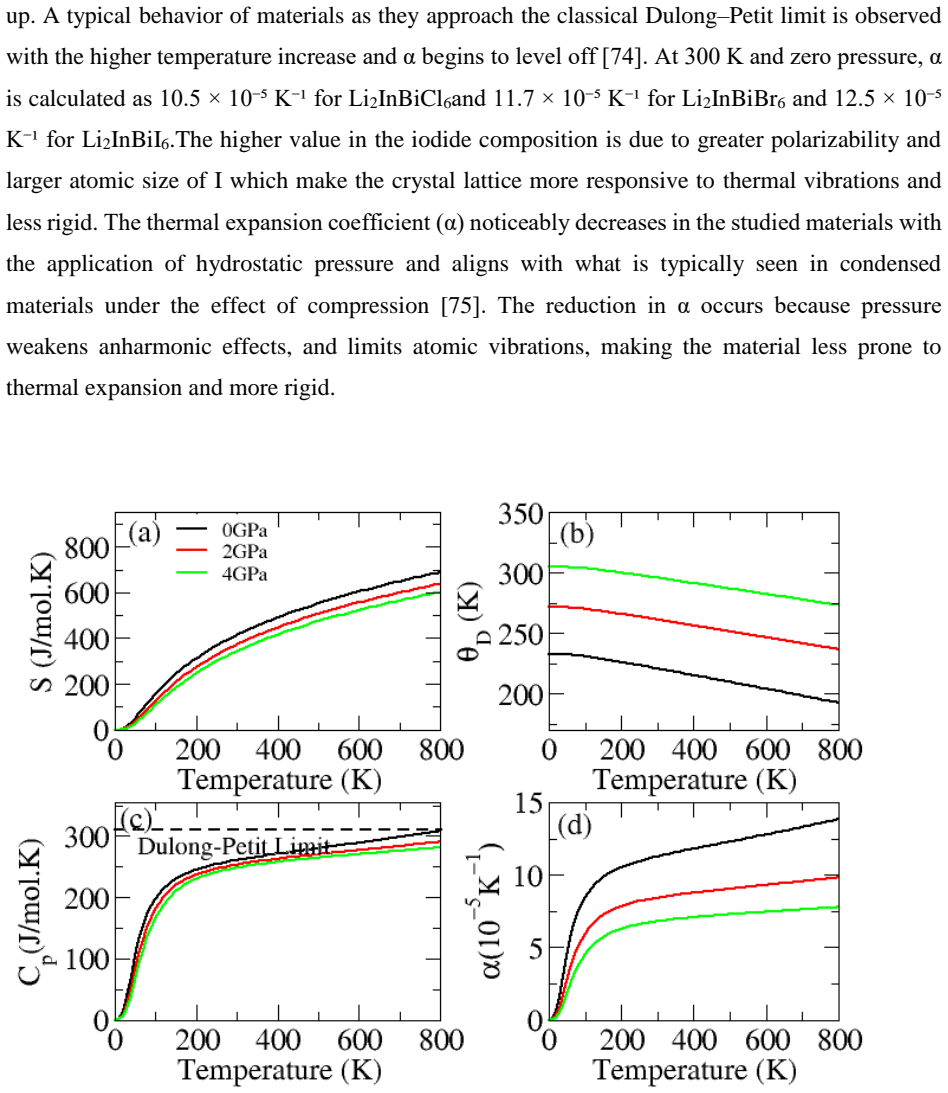
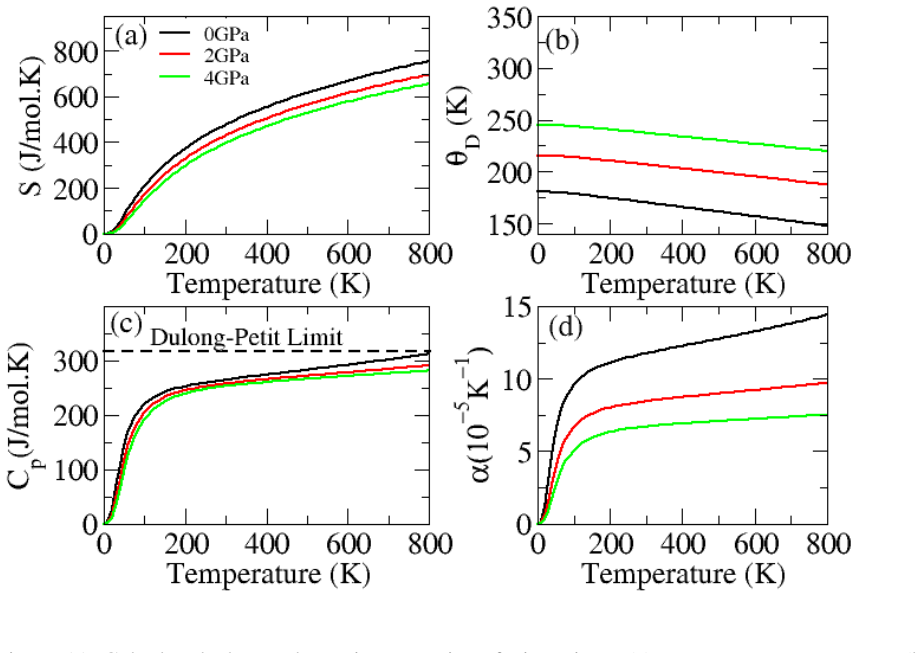
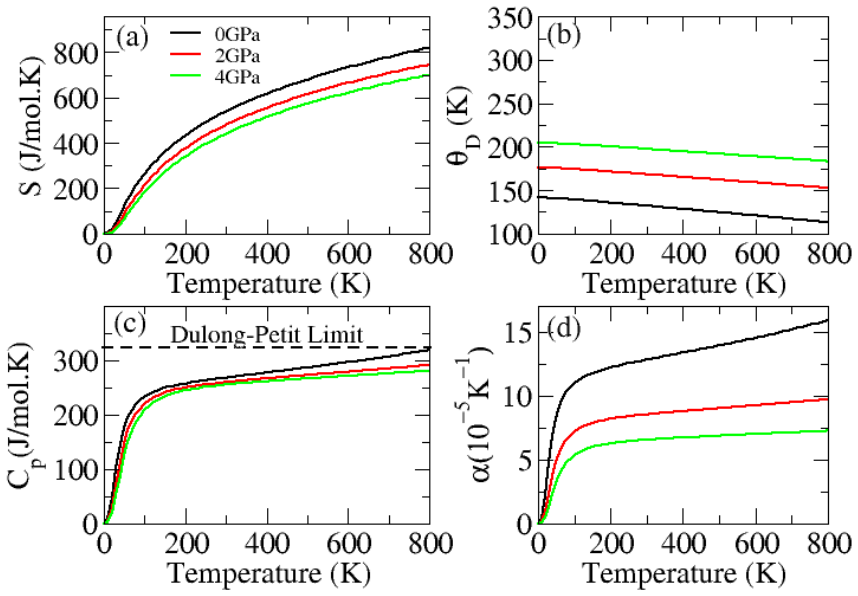
The paper establishes that Li2InBiCl6, Li2InBiBr6, and Li2InBiI6 are cubic double perovskites with formation energies confirming thermodynamic stability. They display direct semiconducting bandgaps of 1.7 eV, 1.3 eV, and 1.1 eV respectively. Analysis of the dielectric function reveals strong optical absorption suitable for optoelectronics, while calculated Seebeck coefficients, conductivities, and ZT values indicate good thermoelectric performance enhanced by the small gaps. The band positions also make them viable for photocatalytic water oxidation across pH 0 to 7.
What carries the argument
The systematic variation of the halogen X in Li2InBiX6 within density functional theory to control the direct bandgap, optical absorption edge, and thermoelectric transport coefficients.
If this is right
- The direct bandgaps and absorption spectra make these halides candidates for photovoltaic and optoelectronic devices.
- The relatively small bandgaps lead to improved power factors and thermoelectric efficiency over the 30-800 K range.
- Band alignment supports their use as photocatalysts for water oxidation in acidic to neutral environments.
- Thermodynamic stability in the cubic phase suggests they can be synthesized and used in practical applications.
Where Pith is reading between the lines
- Replacing chlorine with iodine systematically narrows the gap, offering a way to match specific solar spectrum regions or operating temperatures.
- These materials may offer advantages over lead-based perovskites by avoiding toxicity while maintaining suitable electronic properties.
- Further studies could explore alloying or strain to further optimize the figure of merit or absorption.
Load-bearing premise
The particular density functional and other computational settings used produce bandgaps, optical responses, and transport properties that match the real materials closely enough for the conclusions to hold.
What would settle it
Synthesis of one of the compounds, such as Li2InBiBr6, and direct measurement of its bandgap via absorption spectroscopy or its thermoelectric figure of merit at room temperature to compare against the calculated 1.3 eV and ZT values.
Figures
read the original abstract
Double perovskite halides are emerging as promising materials for a wide range of applications, particularly in renewable energy technologies such as solar cell devices, thereby contributing to addressing global energy demands. In this work, the structural, electronic, optical, dielectric, thermoelectric, and photocatalytic properties of Li2InBiX6 (X = Cl, Br, I) halides are systematically investigated using density functional theory. The calculated formation energies confirm the thermodynamic stability of these compounds in the cubic phase. The studied materials exhibit semiconducting behavior with direct bandgaps of 1.7 eV, 1.3 eV, and 1.1 eV for Li2InBiCl6, Li2InBiBr6, and Li2InBiI6, respectively. The complex dielectric function is analyzed to explore their optical response, revealing strong absorption in the infrared and visible regions, indicating suitability for optoelectronic applications. Thermoelectric properties, including the Seebeck coefficient, electrical conductivity, and figure of merit (ZT), are evaluated over a temperature range of 30 to 800 K. The relatively small bandgaps contribute to enhanced thermoelectric performance, reflected in improved power factors. Furthermore, photocatalytic analysis indicates that Li2InBiX6 compounds are suitable candidates for water oxidation reactions within the pH range of 0 to 7. Overall, the combined thermoelectric and optical performance highlights these double perovskite halides as promising materials for future energy conversion applications.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The paper reports a DFT investigation of the structural, electronic, optical, dielectric, thermoelectric, and photocatalytic properties of the cubic double-perovskite halides Li2InBiX6 (X = Cl, Br, I). It claims thermodynamic stability from negative formation energies, direct bandgaps of 1.7 eV, 1.3 eV, and 1.1 eV respectively, strong absorption in the infrared and visible regions, temperature-dependent thermoelectric quantities (Seebeck coefficient, power factor, ZT) from 30–800 K, and band-edge alignment suitable for photocatalytic water oxidation at pH 0–7, concluding that the materials are promising for optoelectronic and energy-conversion applications.
Significance. If the computed electronic structures and transport coefficients prove accurate, the work would add three new halide compositions to the growing database of double perovskites and illustrate clear halogen-substitution trends in bandgap, absorption, and thermoelectric performance. Such systematic computational surveys can guide experimental synthesis, but only when the underlying DFT methodology is fully documented and validated.
major comments (3)
- [Abstract / Methods] Abstract and (presumed) Methods section: the exchange-correlation functional, Hubbard U (if any), plane-wave cutoff, k-point sampling, and convergence criteria are never stated. Because the reported direct bandgaps (1.7/1.3/1.1 eV) are the sole input to the dielectric-function, absorption, photocatalytic alignment, and Boltzmann-transport calculations, the absence of this information makes it impossible to judge whether the central claims rest on a reliable electronic structure.
- [Results (electronic/optical)] Results (electronic and optical sections): no hybrid-functional, GW, or experimental benchmark is provided to quantify the expected GGA bandgap underestimation (typically 0.5–1 eV for similar halides). Without such a check, the claimed suitability for visible-light absorption and the derived ZT values cannot be considered robust.
- [Thermoelectric properties] Thermoelectric section: the transport calculations lack any description of the relaxation-time approximation, carrier concentration, or code used (BoltzTraP, etc.). These details are load-bearing for the reported power factors and ZT maxima.
minor comments (2)
- [Thermoelectric properties] The temperature range “30 to 800 K” is stated without clarifying whether all thermoelectric quantities are evaluated on the same grid or whether any low-temperature approximations are invoked.
- [Figures] Figure captions and axis labels should explicitly state the functional and smearing parameters used for the dielectric function and density of states.
Simulated Author's Rebuttal
We thank the referee for the careful reading and constructive criticism. The comments highlight important omissions that affect the reproducibility and robustness assessment of our DFT study. We address each major comment below and will revise the manuscript to incorporate the requested information and clarifications.
read point-by-point responses
-
Referee: [Abstract / Methods] Abstract and (presumed) Methods section: the exchange-correlation functional, Hubbard U (if any), plane-wave cutoff, k-point sampling, and convergence criteria are never stated. Because the reported direct bandgaps (1.7/1.3/1.1 eV) are the sole input to the dielectric-function, absorption, photocatalytic alignment, and Boltzmann-transport calculations, the absence of this information makes it impossible to judge whether the central claims rest on a reliable electronic structure.
Authors: We agree that the absence of these computational parameters is a significant oversight that prevents proper evaluation of the results. In the revised manuscript we will insert a dedicated Computational Methods section that explicitly states the exchange-correlation functional, any Hubbard U value (or its absence), the plane-wave cutoff energy, the k-point sampling grid, and all convergence thresholds employed. This addition will allow readers to assess the reliability of the electronic structures used for all subsequent optical, photocatalytic, and transport calculations. revision: yes
-
Referee: [Results (electronic/optical)] Results (electronic and optical sections): no hybrid-functional, GW, or experimental benchmark is provided to quantify the expected GGA bandgap underestimation (typically 0.5–1 eV for similar halides). Without such a check, the claimed suitability for visible-light absorption and the derived ZT values cannot be considered robust.
Authors: We acknowledge the value of benchmarking the GGA bandgaps. Because these specific compositions have not yet been synthesized, no experimental data exist. In the revision we will add an explicit discussion of the known GGA underestimation for halide perovskites, cite hybrid-functional results for chemically related double perovskites, and note the implications for the reported absorption edges and ZT values. If resources permit, we will also perform a limited hybrid-functional calculation on one composition to provide a direct internal benchmark. revision: partial
-
Referee: [Thermoelectric properties] Thermoelectric section: the transport calculations lack any description of the relaxation-time approximation, carrier concentration, or code used (BoltzTraP, etc.). These details are load-bearing for the reported power factors and ZT maxima.
Authors: We agree that these methodological details are essential. The transport properties were obtained from the semi-classical Boltzmann equation under the constant-relaxation-time approximation. In the revised manuscript we will add a concise description of the relaxation-time value adopted, the carrier-concentration range examined, and the code used to solve the transport equation. This will make the origin of the power-factor and ZT curves fully transparent. revision: yes
Circularity Check
No circularity: direct DFT outputs with no self-referential fitting or definitional loops
full rationale
The paper computes structural, electronic (direct bandgaps), optical (dielectric function), thermoelectric (Seebeck, conductivity, ZT), and photocatalytic properties via standard density functional theory. These quantities are obtained as direct numerical outputs from the chosen functional and k-point sampling; none are fitted to the target metrics, redefined in terms of themselves, or justified solely by self-citation. Formation-energy stability checks are independent of the bandgap and transport results. No equations or sections reduce the claimed predictions to the inputs by construction.
Axiom & Free-Parameter Ledger
free parameters (2)
- DFT exchange-correlation functional and Hubbard U (if any)
- k-point sampling density and plane-wave cutoff
axioms (2)
- domain assumption Cubic phase is the thermodynamically stable structure for all three compositions
- domain assumption Boltzmann transport equation within constant relaxation time approximation yields reliable thermoelectric coefficients
Reference graph
Works this paper leans on
-
[1]
https://doi.org/10.1080/14786440808520496. [38]. Munsif, M.; Neffati, R.; Shah, M.; Khan, S.; Waqar Ashraf, M.; Murtaza, G. First Principles Study of the Structural, Mechanical and Optical Properties of Argyrodite - Structured Ag6PS5X (X= Br, I) Compounds. Solid State Commun. 2023, 371, 115245. https://doi.org/10.1016/j.ssc.2023.115245. [39]. Haque, E.; H...
-
[2]
Materials Advances, 2(13), 4277-4290. [69]. Gurunani, B., & Gupta, D. C. (2025). First-principles investigation of thermoelectric performance in KMnZ (Z= Sn, Pb) half-Heusler alloys. RSC Advances, 15(7), 4874-4891. [70]. Johari, G. P. (2021). Entropy, enthalpy and volume of perfect crystals at limiting high pressure and the third law of thermodynamics. Th...
work page 2025
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.