Density Functional Theory Study of Lanthanide Monoxides under High Pressure: Pressure-Induced B1-B2 Transition
Pith reviewed 2026-05-10 16:11 UTC · model grok-4.3
The pith
Density functional theory calculations predict that all fifteen lanthanide monoxides undergo a B1 to B2 structural phase transition under high pressure.
A machine-rendered reading of the paper's core claim, the machinery that carries it, and where it could break.
Core claim
Using density functional theory with the general gradient approximation, the B1 (NaCl-type) structure is the most thermodynamically stable phase at ambient pressure for all fifteen lanthanide monoxides. At elevated pressures every compound undergoes a first-order structural transition to the B2 (CsCl-type) phase. The pressure-volume relation and the isothermal bulk modulus are obtained from the same calculations.
What carries the argument
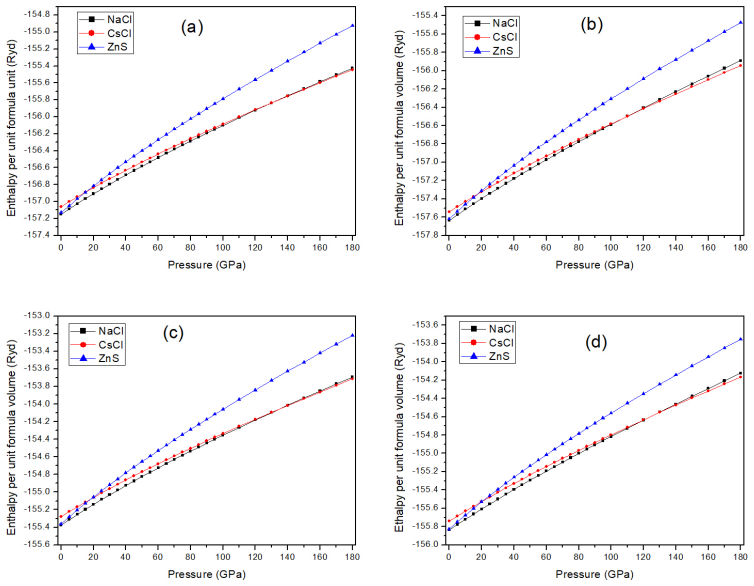
Enthalpy-versus-pressure curves computed for the B1, B2, and B3 cubic structures; the pressure at which the B2 curve crosses below the B1 curve marks the transition point for each compound.
If this is right
- B1 remains the stable structure at zero pressure for the entire lanthanide monoxide series.
- All fifteen compounds share the same B1-to-B2 transition sequence under compression.
- The calculated pressure-volume data furnish an isothermal equation of state and bulk modulus for each material.
- GGA reproduces experimental lattice parameters and volumes of the B1 phase more closely than LDA.
Where Pith is reading between the lines
- The predicted transition pressures could serve as targets for future diamond-anvil-cell experiments on these compounds.
- Because the study treats the full lanthanide series uniformly, trends in transition pressure or bulk modulus across the row become directly comparable.
- The same computational protocol could be extended to related rare-earth compounds to test whether the B1-B2 sequence is a general feature under high pressure.
Load-bearing premise
The assumption that the general gradient approximation supplies accurate enough total energies and pressures to locate the B1-B2 crossing correctly.
What would settle it
Direct experimental measurement of the pressure at which each lanthanide monoxide transforms from B1 to B2; systematic mismatch between measured and calculated transition pressures would falsify the predictions.
Figures



read the original abstract
Using density functional theory, we study the influence of hydrostatic pressure on the crystal structure of lanthanide monoxides, focusing on the monoxides formed by the fifteen elements of the lanthanide series, from La to Lu. Calculations are performed using two methods for the ambient pressure B1 (NaCl type) structure, the general gradient approximation (GGA) and the local density approximation (LDA). Through a systematic comparison with existent experimental data, we find that the first method agrees better with the experiments. In addition, considering other cubic structures previously reported for lanthanide monoxides, as B2 (CsCl type) and B3 (ZnS type), we explore the possibility of the occurrence of pressure-induced phase transitions. Based on the better accuracy of GGA to describe the B1 phase at ambient conditions, we exclusively use GGA for the high pressure study. We find, for the fifteen studied compounds, that, at ambient pressure, the B1 structure is the one with the lowest enthalpy, being therefore the most thermodynamically stable structure. We also determine that, at elevated pressures, all the studied compounds undergo a structural phase transition to the B2 phase. We finally establish the relationship between pressure and volume of the unit cell, along with the associated isothermal equation of state, determining the bulk modulus.
Editorial analysis
A structured set of objections, weighed in public.
Referee Report
Summary. The manuscript uses DFT to examine lanthanide monoxides (LaO to LuO) in the B1 (NaCl-type) structure at ambient pressure, comparing GGA and LDA results to experiment and finding GGA superior for lattice constants and bulk moduli. It then applies GGA to explore high-pressure behavior, reporting that B1 is the lowest-enthalpy phase at ambient conditions for all fifteen compounds while B2 (CsCl-type) becomes favored at elevated pressures; B3 (ZnS-type) is considered but not competitive. The work also presents the pressure-volume equation of state and associated bulk moduli.
Significance. If the GGA-based transition pressures hold, the study supplies a systematic, element-by-element prediction of a universal B1-to-B2 transition across the entire lanthanide monoxide series. This could serve as a useful guide for future high-pressure experiments on f-electron materials and for modeling their behavior under compression. The ambient-pressure validation against existing experimental data for the B1 phase provides a concrete benchmark that strengthens the baseline calculations.
major comments (2)
- Abstract and high-pressure study: the decision to restrict the high-pressure analysis to GGA is justified solely by its better agreement with ambient-pressure B1 data, yet this does not test whether the same functional remains reliable for B1-B2 enthalpy crossovers once volumes are reduced, where 4f hybridization and correlation effects may shift.
- Computational details section: the manuscript omits essential parameters such as k-point sampling densities, plane-wave cutoffs, pseudopotential choices, and convergence criteria for energy and forces. Without these, the numerical accuracy of the reported transition pressures and bulk moduli cannot be assessed or reproduced.
minor comments (1)
- The presentation would be improved by adding uncertainty estimates or sensitivity tests on the computed transition pressures to indicate how robust the universal B1-B2 claim is to small variations in computational settings.
Simulated Author's Rebuttal
We thank the referee for the detailed and constructive report. We address each major comment below and indicate the revisions made to the manuscript.
read point-by-point responses
-
Referee: Abstract and high-pressure study: the decision to restrict the high-pressure analysis to GGA is justified solely by its better agreement with ambient-pressure B1 data, yet this does not test whether the same functional remains reliable for B1-B2 enthalpy crossovers once volumes are reduced, where 4f hybridization and correlation effects may shift.
Authors: We acknowledge that the justification for employing GGA rests on its improved description of the ambient-pressure B1 phase relative to LDA. While GGA is routinely applied to high-pressure structural transitions in f-electron systems, we agree that its performance for enthalpy crossovers at compressed volumes is not independently validated here. In the revised manuscript we have added a dedicated paragraph in the Discussion section that explicitly notes this limitation, states that the reported transition pressures should be regarded as indicative, and suggests that future studies incorporating DFT+U or hybrid functionals would be valuable to assess possible shifts arising from enhanced 4f hybridization. revision: partial
-
Referee: Computational details section: the manuscript omits essential parameters such as k-point sampling densities, plane-wave cutoffs, pseudopotential choices, and convergence criteria for energy and forces. Without these, the numerical accuracy of the reported transition pressures and bulk moduli cannot be assessed or reproduced.
Authors: We thank the referee for identifying this omission. The revised manuscript now contains an expanded Computational Methods section that specifies all required parameters: a plane-wave cutoff of 520 eV, Monkhorst-Pack k-point meshes with a reciprocal-space density of 0.025 Å⁻¹, projector-augmented-wave pseudopotentials, and convergence thresholds of 10^{-6} eV for total energy and 0.01 eV/Å for forces. These settings were used uniformly for all enthalpy and equation-of-state calculations. revision: yes
Circularity Check
No circularity: standard DFT validation against external ambient data followed by direct high-pressure enthalpy computation
full rationale
The paper computes total energies and enthalpies for B1, B2, and B3 structures using standard GGA and LDA functionals in a DFT code. Ambient-pressure lattice constants and bulk moduli are compared to independent experimental literature values to select GGA; high-pressure B1-B2 crossovers are then obtained by direct minimization of H(P) = E(V) + PV for each compound. No parameters are fitted to the transition pressures themselves, no self-citations supply the central result, and the derivation does not reduce any claimed prediction to a redefinition or renormalization of its own inputs. The chain is therefore self-contained against external benchmarks.
Axiom & Free-Parameter Ledger
axioms (1)
- domain assumption Density functional theory with GGA sufficiently approximates exchange-correlation effects to predict relative enthalpies and phase stability in lanthanide monoxides under pressure.
Reference graph
Works this paper leans on
-
[1]
E. G. Rauh and R. J. Ackermann, The first ionization potentials of neptunium and neptunium monoxide, J. Chem. Phys. 62 (1975) 1584
work page 1975
-
[2]
E. Murad and D. L. Hildenbrand, Dissociation energies of GdO, HoO, ErO, TmO, and LuO; correlation of results for the lanthanide monoxide series, J. Chem. Phys. 73 (1980) 4005 - 4011. 9
work page 1980
-
[4]
H. Gan, C. Zhang, X. Z. Du, P. Jiang, C. P. Niu, X. H. Zheng, Y . W. Yin, and X. G. Li, Insights into superconductivity of LaO from experiments and first-principles calculations, Phys. Rev. B 104 (2021) 054515
work page 2021
- [5]
- [7]
-
[8]
P. H. Sun, J. F. Zhang, K. Liu, Q. Han, and Z. Y . Lu, First -principles study of the superconductivity in LaO, Phys. Rev. B 104 (2021) 045121
work page 2021
-
[9]
K. Kaminaga, D. Oka, T. Hasegawa, and T. Fukumura, Superconductivity of rock-salt structure LaO epitaxial thin film, J. Am. Chem. Soc. 140 (2018) 6754-6757
work page 2018
-
[10]
M. Jurkutat, C. Kattinger, S. Tsankov, and J. Haase, How pressure enhances the critical temperature of superconductivity in YBa2Cu3O6+y, PNAS 120 (2023) e22154581
work page 2023
-
[11]
C, X, Chi, H. Xie, R. Cong, Z, C. Tang, and M. F. Zhou, Electron Affinities of the Early Lanthanide Monoxide Molecules, Chin. J. Chem. Phys. 24 (2011) 604-610
work page 2011
-
[12]
S. S. Harilal, E. J. Kautz, B. E. Bernacki, M. C. Phillips, P. J. Skrodzki, M. Burger, and I. Jovanovic, Physical conditions for UO formation in laser -produced uranium plumes, Phys. Chem. Chem. Phys. 21 (2019) 16161-16169
work page 2019
-
[13]
P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. Fabris, G. Fratesi, S. de Gironcoli, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin- Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Sc...
work page 2009
-
[14]
J. P. Perdew, K, Burke, and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett. 77 (1996) 3865
work page 1996
- [15]
-
[16]
J. P. Attfiled and G. Ferey, Structural correlations within the lanthanum palladium oxide family, J. Sol. State Chem. 80 (1989) 286-298
work page 1989
-
[17]
A. B. Garg, A. Muñoz, S. Anzellini, J. Sanchez -Martín, R. Turnbull, D. Díaz - Anichtchenko, C. Popescu, and D. Errandonea, Role of GdO addition in the structural stability of cubic Gd2O3 at high pressures: Determination of the equation of states of GdO and Gd2O3, Materialia 34 (2024) 102064
work page 2024
-
[18]
H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B 13 (1976) 5188-5192
work page 1976
-
[19]
Birch, Finite Elastic Strain of Cubic Crystals, Phys
F. Birch, Finite Elastic Strain of Cubic Crystals, Phys. Rev. 71 (1947) 809-824
work page 1947
-
[20]
J. M. Leger, N. Yacoubi, and J. Loriers, Synthesis of rare earth monoxides, J. Sol. State Chem. 36 (1981) 261-270
work page 1981
-
[21]
S. D. Griesemer, L. Ward, and C. Wolverton, High- throughput crystal structure solution using prototypes, Phys. Rev. Mater. 5 (2021) 105003
work page 2021
-
[22]
J. M. Leger, N. Yacoubi, and J. Loriers, Mater. Res. Bull. 14, 1431 (1979)
work page 1979
-
[23]
G. Krill, M.F. Ravet, J.P. Kappler, L. Abadli, J.M. Leger, N. Yacoubi, and C. Loriers, Magnetic properties of some rare earth monoxides LnO (Ln = Pr, Nd, Sm) mixed valence state of SmO, Sol. State Comm. 33 (1980) 351-353
work page 1980
-
[24]
S. Kobayashi, A. Martín-Cid, K. Toyoki, H. Okazaki, S. Hirosawa, and T. Nakamura, Influence of magnetostriction on the lattice constants of the secondary phases in Nd -Fe- B sintered magnets studied by synchrotron X -ray diffraction, AIP Advances 9 (2019) 25154
work page 2019
-
[25]
H. A. Eick, N. C. Baezinger, and L. Eyring, Lower oxides of samarium and europium. The preparation and crystal structure of SmO0.4-0.6, SmO and EuO, J. Am. Chem. Soc. 78 (1956) 5147-5149
work page 1956
-
[26]
J. M. Leger, P. Aimonino, J. Loriers, P. Dordor, and B Coqblin, Transport properties of SmO, Phys. Lett. A 80 (1980) 325-327
work page 1980
-
[27]
B. T. Matthias, R. M. Bozorth and J. H. Van Vleck, Ferromagnetic Interaction in EuO, Phys. Rev. Lett. 7 (1961) 160-161. 11
work page 1961
-
[28]
M. Namba, H. Takatsu, R. Mikita, Y . Sijia, K. Murayama, H.- B. Li, R. Terada, C. Tassel, H. Ubukata, M. Ochi, R. Saez-Puche, E. Palacios Latasa, N. Ishimatsu, D. Shiga, H. Kumigashira, K. Kinjo, S. Kitagawa, K. Ishida, T. Terashima, K. Fujita, T. Mashiko, K. Yanagisawa, K. Kimoto, and H. Kageyama, Large perpendicular magnetic anisotropy induced by an int...
work page 2023
-
[29]
T. Yamamoto, K. Kaminaga, D. Saito, D. Oka, and T. Fukumura, Rock salt structure GdO epitaxial thin film with a high ferromagnetic Curie temperature, Appl. Phys. Lett. 117 (2020) 052402
work page 2020
- [30]
-
[31]
T. Amrillah, D. Oka, H. Shimizu, S. Sasaki, D. Saito, K. Kaminaga, and T. Fukumura, Rock salt-type HoO epitaxial thin film as a heavy rare- earth monoxide ferromagnetic semiconductor with a Curie temperature above 130 K, Appl. Phys. Lett. 120 (2022) 082403
work page 2022
-
[32]
J. M. Leger, J. Maugrion, L. Albert, J. C. Achard, and C. Lories, High pressure synthesis of ytterbium oxides (YbO and Yb 3O4), C. R. Acad. Sci. Serie C, Sciences Chimiques 286, 201 (1978)
work page 1978
-
[33]
N.A. Fishel, J.M. Haschke, and H.A. Eick, Preparation of ytterbium and europium oxides in liquid ammonia, Inor. Chem. 9 (1970) 413-414
work page 1970
-
[34]
J. M. Leger, N. Yacoubi, J. Loriers, G.J. McCarthy, J.J. Rhyne, and H.B. Silber (Eds.), The rare earths in modern science and technology, V ol. 2, Plenum Press, New York and London (1979), p. 203
work page 1979
-
[35]
G. X. Zhang, A. M. Reilly, A. Tkatchenko, and M. Scheffler, Performance of various density-functional approximations for cohesive properties of 64 bulk solids, New J. Phys. 20 (2018) 063020
work page 2018
-
[36]
P. Söderlind, P. E. A. Turchi, A. Landa, and V . Lordi, Ground-state properties of rare- earth metals: an evaluation of density-functional theory, J. Phys.: Cond. Matter 26 (2014) 416001. [37] K. A. Gschneidner Jr., Systematics in lanthanide and actinide solids, J. Alloys Compd. 223 (1995) 165-169
work page 2014
- [37]
-
[38]
Johansson, Nature of the electrons in the actinide series, Phys
B. Johansson, Nature of the electrons in the actinide series, Phys. Rev. B 11, 2740 (1975)
work page 1975
-
[39]
G. Kalpana, B. Palanivel, and M. Rajagopalan, Electronic and structural properties of alkaline-earth oxides under high pressure, Phys. Rev. B 52, (1995) 4-7
work page 1995
-
[40]
H. Liu, H. K. Mao, M. Somayazulu, Y . Ding, Y . Meng, and D. Häusermann, B1-to- B2 phase transition of transition- metal monoxide under strong compression, Phys. Rev. B 70 (2004) 094114
work page 2004
-
[41]
C. E. Sims, G. D. Barrera, N. L. Allan, and W. C. Mackrodt, Thermodynamics and mechanism of the B1 -B2 phase transition in group- I halides and group- II oxides, Phys. Rev. B 57 (1998) 11164
work page 1998
-
[42]
P. Richet, H.K. Mao, P.M. Bell, Static compression and equation of state of CaO to 1.35 Mbar, J. Geophys. Res. 93(1988) 15279-15288
work page 1988
-
[43]
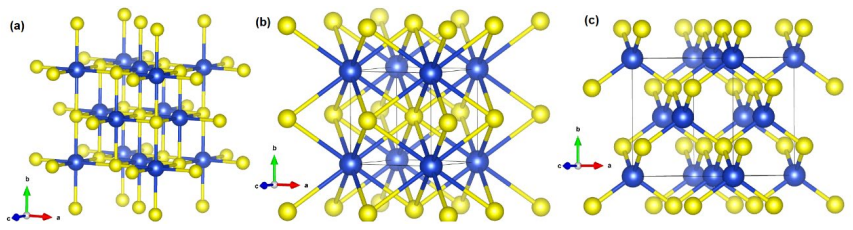
H. K. Mao and P. M. Bell, Equations of state of MgO and ε Fe under static pressure conditions, J. Geophys. Res. 84 (1979) 4533-4536. 13 Figure 1: Crystal structure of the (a) B1, (b) B2, and (B3) phases. The blue (yellow) spheres represent the lanthanide (oxygen) atoms. 14 Figure 2: Enthalpy versus pressure for (a) LaO, (b) CeO, (c) PrO, and (d) NdO. 15 F...
work page 1979
discussion (0)
Sign in with ORCID, Apple, or X to comment. Anyone can read and Pith papers without signing in.